

Abstract
Kufs disease, an adult-onset neuronal ceroid lipofuscinosis, is challenging to diagnose and genetically heterogeneous. Mutations in CLN6 were recently identified in recessive Kufs disease presenting as progressive myoclonus epilepsy (Type A), whereas the molecular basis of cases presenting with dementia and motor features (Type B) is unknown. We performed genome-wide linkage mapping of two families with recessive Type B Kufs disease and identified a single region on chromosome 11 to which both families showed linkage. Exome sequencing of five samples from the two families identified homozygous and compound heterozygous missense mutations in CTSF within this linkage region. We subsequently sequenced CTSF in 22 unrelated individuals with suspected recessive Kufs disease, and identified an additional patient with compound heterozygous mutations. CTSF encodes cathepsin F, a lysosomal cysteine protease, dysfunction of which is a highly plausible candidate mechanism for a storage disorder like ceroid lipofuscinosis. In silico modeling suggested the missense mutations would alter protein structure and function. Moreover, re-examination of a previously published mouse knockout of Ctsf shows that it recapitulates the light and electron-microscopic pathological features of Kufs disease. Although CTSF mutations account for a minority of cases of type B Kufs, CTSF screening should be considered in cases with early-onset dementia and may avoid the need for invasive biopsies.
INTRODUCTION
The neuronal ceroid lipofuscinoses (NCLs) are neurodegenerative diseases characterized by the abnormal accumulation of lipopigment in lysosomes. Kufs disease, the most common adult form, is a challenging diagnosis; the symptoms can be due to a variety of other disorders and pathological diagnosis may require brain biopsy. Even then, it may require specialized expertise to distinguish abnormal ceroid-lipofuscin from normal age-pigment and diagnostic doubt may remain.
Diagnostic difficulty has delayed discovery of the molecular defects underlying adult NCLs, which has lagged behind that of the childhood onset forms. The first genes implicated in Kufs disease were reported in 2011. We previously reported that mutations in CLN6 (MIM 606725, http://www.omim.org) cause recessive Type A Kufs disease (1) (MIM 204300) that typically presents with progressive myoclonus epilepsy. Mutations in DNAJC5 (MIM 611203) have been found in some cases of dominant Kufs (2–4) (MIM 162350), also presenting with progressive myoclonus epilepsy.
Here we analyzed unsolved cases from our earlier study with Type B Kufs disease, where dementia and motor disturbances, rather than epilepsy, dominate the clinical picture. We targeted two well-characterized informative recessive Type B families (Ku4, Ku10; Fig. 1), analyzing them with linkage mapping followed by exome sequencing. Following the identification of the disease-causing locus and putative gene, we then extended the screening to a variety of unsolved cases.
Figure 1.

Kufs disease pedigrees and CTSF mutational status. Individuals from Ku4 and Ku10 whose genotypes were used for linkage analysis are indicated with a hash sign. Individuals whose exomes were sequenced are indicated with an asterisk.
RESULTS
Linkage mapping
We targeted two well-characterized families showing recessive inheritance of Type B Kufs disease: Italian family Ku4 and French-Canadian family Ku10 (Figs 1 and 2). We have previously shown that the three patients from these families do not have mutations in CLN6 (1) (see Materials and Methods for clinical details). As part of this previous study, linkage mapping was performed for Ku4, but not for Ku10. For the current study, we genotyped the Ku10 proband and four unaffected siblings. The parents of Ku10 were reported to be second cousins, and this relationship was verified using FEstim analysis (5); estimated inbreeding coefficients for the children ranged between 0.009 and 0.023 (median 0.018) compared with an expected value of 0.016.
Figure 2.
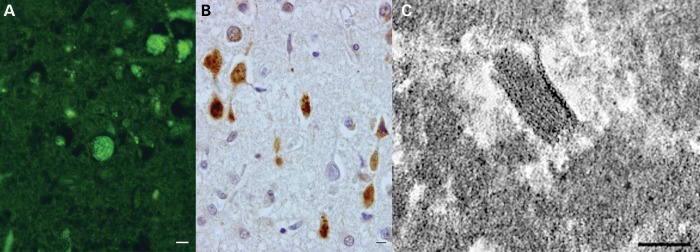
Brain pathology of the proband (II-3) of family Ku4. Light microscopy (A and B: at same magnification; bar = 25 µm) shows cortical neurons containing abundant storage material that is autofluorescent (A, yellow) and immunoreactive for ubiquitin (B, brown reaction product; anti-ubiquitin polyclonal antibody, 1:1000; DakoCytomation). Note anti-ubiquitin immunolabeling extending to swollen proximal axons, a feature characteristic of NCL. Electron microscopy (C; bar = 100 nm) shows diagnostic fingerprint profiles in neurons.
Linkage mapping of Ku10 identified a unique linkage peak achieving a maximum LOD score of 2.30 on chromosome 11 (Fig. 3). This peak, located between SNP markers rs10767218 and rs7124728, extended over 36.7 cM (46.21 Mb) and contained 868 genes. We noticed that this peak was entirely overlapped by one of the 18 linkage peaks previously detected in Ku4 (1) (Fig. 3), suggesting that the same gene might be mutated in both families. Ku4 is a small non-consanguineous family, with the maximum LOD score achieved being 0.86 (1).
Figure 3.

Overlap of linkage peaks obtained by linkage analysis of Ku10 and Ku4. Genome-wide LOD scores were obtained by linkage analysis of the Ku4 and Ku10 families under a fully penetrant recessive genetic model.
Exome sequencing
We sequenced the exomes of the mother and her affected daughter in Ku10, and of the father and his two affected daughters in Ku4. We detected 3837 variants within the unique Ku10 linkage peak in mother I-2 and/or daughter II-1. Of these, 386 variants were rare, being absent from or having alternate allele frequencies of <1% in each of three variant databases: the NHLBI Exome Sequencing Project (ESP6500) database (6503 exomes, http://evs.gs.washington.edu/EVS/), version 2 of the May 2011 release of the 1000 Genomes Project (6) (1092 individuals) and an in-house database (123 exomes). Eighteen of these variants were predicted to affect the amino acid sequence or splicing of the resulting protein; of these, two had genotypes consistent with the recessive consanguineous disease model (i.e. affected daughter homozygous and unaffected mother heterozygous). These two variants were a non-synonymous change in CTSF, which was absent from all databases examined, and a non-synonymous change in CPT1A (MIM 600528) that was present in dbSNP135 (http://www.ncbi.nlm.nih.gov/projects/SNP/) and the two public databases (Supplementary Material, Table S3).
Mutations in CPT1A are known to cause carnitine palmitoyltransferase 1A deficiency (MIM 255120), while mutations in CTSF have not yet been associated with any phenotype in humans. As CTSF encodes the lysosomal cysteine protease cathepsin F, the variant in this gene appeared most biologically plausible. The variant was a c.962A > G (p.Gln321Arg) change in exon 7 (RefSeq accession number NM_003793.3). Sanger sequencing confirmed that the variant was genuine (Supplementary Material, Fig. S1) and segregated as expected in the family (Fig. 1), with the mother and father heterozygous for the variant and none of five unaffected siblings homozygous for the variant.
Exome sequencing in the Ku4 family detected two additional novel variants in CTSF and no rare coding or splice site variants in CPT1A. Both affected siblings in Ku4 were predicted to be heterozygous for a c.1373G > C (p.Gly458Ala) variant in exon 12 of CTSF, as was their father. Affected sibling II-2 was predicted to be homozygous for a second variant in this gene, c.1439C > T (p.Ser480Leu) in exon 13, while her sister and father were predicted to be homozygous for the reference allele, with the variant location covered by just two reads for each of the three samples. Sanger sequencing demonstrated that both affected siblings were in fact heterozygous for both variants, and that the variants segregated as expected in the family (Fig. 1). No DNA sample was available from the deceased mother, so we could not verify that she was heterozygous for the p.Ser480Leu change as expected. Sequence traces for the two variants are shown in Supplementary Material, Fig. S1.
Screening for CTSF mutations in other cases and in controls
We subsequently sequenced CTSF in 22 unrelated individuals. The sample comprised nine unrelated sporadic or putatively recessive cases with pathologically confirmed Kufs disease, three with dominant inheritance of definite or suspected Kufs, and 10 with possible but pathologically unconfirmed Kufs disease (see Supplementary Material, Table S4).
CTSF mutations were found in one patient (Ku16), a 41-year-old Australian woman of British ancestry with clinically suspected, but pathologically unconfirmed, Kufs type B (Fig. 1; see Materials and Methods for clinical details). She was found to be heterozygous for two CTSF variants: a paternally inherited c.962A > G (p.Tyr231Cys) substitution in exon 5 (rs143889283) and a maternally inherited single nucleotide deletion in exon 7 (c.954delC [p.Ser319Leufs*27]) that results in a frameshift and subsequent nonsense mutation (Supplementary Material, Fig. S1). Neither variant is present in dbSNP 135 or the 1000 Genomes databases. The deletion is absent from the ESP 6500 database, while the substitution is present in the heterozygous state in a single individual out of 4295 Americans with European ancestry (minor allele frequency 0.012%) and was not observed in 2200 Americans of African ancestry.
We additionally sequenced CTSF in 376 control chromosomes from blood bank donors. None of the five rare variants found in Kufs Type B patients were detected in the controls, and we did not detect any additional novel variants in CTSF.
In silico modeling
Of the five rare CTSF variants, four (p.Gln321Arg, p.Gly458Ala, p.Ser480Leu and p.Ser319Leufs*27) are located in the peptidase C1 domain towards the C-terminal end of CTSF. The fifth, p.Tyr231Cys, is located within the I29 propeptide inhibitor domain. The four missense mutations affect highly conserved amino acids and are predicted to be damaging by SIFT and ‘probably damaging’ by PolyPhen-2 (Supplementary Material, Table S5), while the single nucleotide frameshift deletion results in a nonsense mutation that truncates the protein to approximately three-quarters of its usual length of 484 amino acids.
The natural substrate for cathepsin F is not known, so alteration of function could not be tested. However, the X-ray structure of human cathepsin F has been solved (7) and served as the basis for in silico modeling. This modeling suggests that the p.Gln321Arg mutation results in a conformational change in the binding loop that would lower catalytic efficiency (Fig. 4). The backbone dihedral angles (Phi/Psi) of Gly458 are in an energetically unfavorable region, which only allows glycine residues. Therefore, the p.Gly458Ala mutation will likely lead to a conformational change or even complete misfolding. Similarly, the side chain of Ser480 is part of an extensive hydrogen bond network, including the side chains of Asn305 and Gln309, in the core of the protein. Disruption of this network by introducing a hydrophobic side chain with the p.Ser480Leu mutation may also lead to misfolding of the enzyme.
Figure 4.
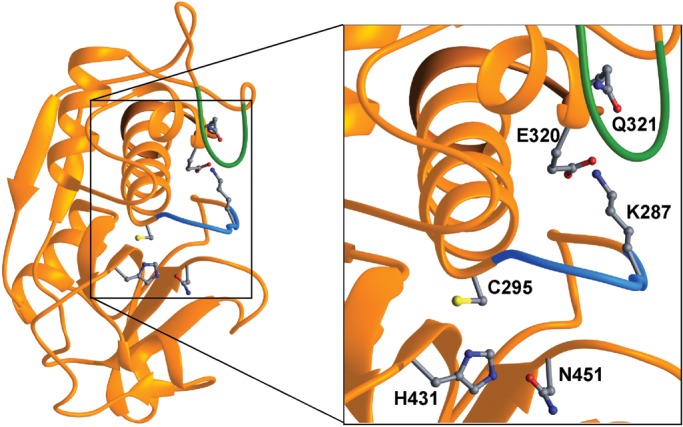
A ribbon representation of the cathepsin F structure (7). Side chains of the catalytic triad (C295, H431 and N451) are depicted. Substrate binding and recognition is mediated by the loop preceding the catalytic C295 (blue). Lysine 287 (K287) is part of this loop and forms an ion pair with glutamic acid 320 (E320). This interaction keeps the loop (blue) in its conformation. The mutated residue Q321 (p.Gln321Arg) is buried under a loop (green). Changing the electrostatic environment by the Gln to Arg mutation at position 321 may lead to disruption of the ion pair E320–K287 and induction of a conformational change in the binding loop that renders substrate binding and lowers the catalytic efficiency.
Pathology of Ctsf-/- mice
Ctsf knockout mice generated by homologous recombination developed signs of neurological disease at 12–16 months of age (8). Histology showed excess accumulation of autofluorescent granules in cerebral cortex, hypothalamus, Purkinje cells and anterior horn cells. Storage of mitochondrial ATPase subunit c could not be demonstrated by immunoblotting.
We obtained high power electron micrographs from anterior horn cells of these same animals, and verified the presence of membrane-bound accumulations of rectilinear complex (Fig. 5), a storage pattern that is seen along with fingerprint profiles in several forms of NCL. This storage was accompanied by cortical atrophy and localized glial activation, characteristic features that are evident in mouse models of multiple forms of NCL (see Supplementary Material, Fig. S2 and Supplementary Text).
Figure 5.

This electron micrograph from an anterior horn cell of a Ctsf knockout mouse shows part of an intracellular membrane-bound organelle filled with rectilinear complex. Pentalaminar structures are present.
DISCUSSION
In total, five rare or novel variants were detected in CTSF (Supplementary Material, Table S6). Familial segregation of these variants is shown in Fig. 1.
Multiple lines of evidence suggest that the variants detected in CTSF are pathogenic mutations. First, they are rare; four are absent from databases containing genotypes from a total of 7618 individuals, while the fifth was present in a heterozygous state in a single individual. Secondly, they are highly likely to have deleterious effects on protein function. Thirdly, in silico modeling of the p.Gln321Arg mutation suggested that it would lower catalytic efficiency. Fourthly, other members of the cathepsin family have been implicated in other forms of NCL and lysosomal storage disorders: mutations in CTSD are known to cause congenital neuronal ceroid lipofuscinosis (9) (MIM 610127), while reduced levels of cathepsin H have been reported in juvenile-onset NCL (MIM 204200) (10), and mutations in CTSA cause galactosialidosis (MIM 256540) (11). Although the natural substrate of cathepsin F is unknown, and the nature of the storage in Kufs disease incompletely characterized, mutation in a cysteine protease like cathepsin F is a very plausible candidate mechanism for this lysosomal storage disease.
Finally, mice deficient in cathepsin F have neurological disease beginning in mid-life (12–16 months) with incoordination and weakness and premature death, and display storage material with the characteristic light and electron microscopic features seen in NCL (8). Our characterization of these mice (Fig. 4, Supplementary Text and Supplementary Material, Fig. S2) confirms that CTSF deficiency in mice also results in an NCL-like disorder. It will be important to define the onset and progression of these phenotypes, information that can be used to judge the efficacy of future therapeutic interventions.
Our experience suggests that most, if not all, cases of recessive Kufs type A are due to mutations in CLN6, although this conclusion awaits confirmation by others. In contrast, CTSF mutations appear to account for a lesser proportion of recessive Kufs type B cases. Kufs Type B is a particularly challenging clinicopathological diagnosis, highlighting the need for molecular diagnosis. Our experience of a modest yield of CTSF mutations is mirrored in an earlier study (8). Following discovery of an NCL phenotype in cathepsin F deficient mice, they sequenced CTSF in 13 patients with suspected late-onset NCL, but did not find any likely pathogenic variants (8). However, these yields are likely spuriously low due to inclusion of diagnostically dubious cases.
We propose that CTSF should receive the locus designation CLN13. Together with the findings that mutations in CLN6 cause recessive Kufs disease Type A and that mutations in DNAJC5 are a cause of dominant Kufs disease, our discovery of the role of CTSF mutations in Kufs Type B disease advances knowledge regarding the molecular basis of Kufs disease. This knowledge will allow some suspected cases of Kufs disease to be confirmed by simple sequencing, as opposed to requiring an invasive brain biopsy. However, as we identified cases of Kufs disease without mutations in either CLN6 or CTSF, additional loci for recessive Kufs disease remain to be discovered.
MATERIALS AND METHODS
Families and case histories
Family Ku4
This Italian family has two affected children and no known consanguinity. The proband was a University student who developed a progressive cerebellar syndrome from age 20, initially manifesting as tremor with subsequent frank ataxia and dysarthria. Rare tonic–clonic seizures occurred from age 28. Progressive cognitive impairment with emotional lability began at around age 30. Her IQ was 71 at age 30 and worsening dementia subsequently evolved. Her vision was preserved. MRI at age 30 showed cortical and cerebellar atrophy. A polygraphic study at age 31 showed that her movements were due to tremor and not myoclonus. There was no epileptiform activity. She died at age 42 years. The neuropathological study of the brain taken at autopsy revealed diffuse cerebral atrophy, with neuronal loss and astrogliosis most severe in the cerebral cortex and in the cerebellum. Abundant autofluorescent material was found in the cytoplasm of neurons of the cerebral cortex, thalamus, striatum, brainstem nuclei and of Purkinje cells. This storage material was immunoreactive for ubiquitin and contained fingerprint profiles at the electron microscopic examination (Fig. 2).
Her older sister developed depression with cognitive decline at age 32. She had rare seizures from age 41. From age 42, ataxia and dysarthria were noted and subsequently pyramidal and extra-pyramidal motor features were seen. Progressive dementia and motor disability ensued and she became bedridden at age 51 years and required a gastrostomy for nutrition at age 54. MRI showed diffuse cerebral atrophy. An EEG did not show epileptiform activity, and a skin biopsy was unremarkable.
There were two healthy sisters, the parents were unrelated and no other family members were known to be affected. Linkage mapping of this family, performed as part of our earlier study (1) with an autosomal recessive genetic model, identified 18 linkage peaks achieving a maximum LOD score of 0.86. The inbreeding coefficient was estimated to be zero for all family members, suggesting a likely compound heterozygous disease model.
Family Ku10
The proband of this French-Canadian family was a businesswoman and talented dancer. From age 24 years she had very occasional focal seizures with minimal impact on her life. From age 35 she had progressive dementia with mood disturbance and motor features including tremor, ataxia, and extra-pyramidal type rigidity with mild hypereflexia.
Institutional care was necessary from age 40 years due to dementia. She required a wheelchair from age 47 and remained alive at age 53 years. Seizures were controlled, there was no generalized myoclonus but some facial jerking was present. Her vision remained preserved.
MRI at age 38 years showed diffuse atrophy. Brain biopsy at age 38 years by light microscopy showed definite autofluorescent, Luxol Fast Blue positive storage material in neurons. The original electron microscopy (EM) blocks were unavailable for examination; the small number of available electron micrographs did not show definite storage in the neurons.
We initially classified her as Kufs disease of uncertain type (1), but further follow-up and review of old records showed that the main clinical features were dementia and motor disturbances; thus the classification was revised to Kufs type B. She had five healthy siblings (Fig. 1) and her parents were known to be second cousins. An extensive pedigree (data not shown) revealed no other affected subjects.
Family Ku16
The proband from this Australian family was a manual worker in steady employment who developed cognitive decline from age 35 years. Initially this was manifest as impaired organization of her home and work duties. She also developed a mild dysarthria. She had a tonic–clonic seizure following a day-long period of confusion at age 39 years. Examination at age 40 years revealed that she was disinhibited and distractable. She had a mild cerebellar dysarthria and mild gait ataxia with tremor and past-pointing; no further seizures occurred.
MRI showed ventricular enlargement and frontal and parietal cortical atrophy. EEG did not show epileptiform activity. A skin biopsy did not have specific diagnostic features; brain biopsy was not performed. Her parents were unrelated, she had two healthy siblings and there was no family history of a similar illness.
DNA extraction and linkage mapping
High-molecular-weight DNA was extracted from peripheral blood cells, skin fibroblasts or stored autopsy material and used for mapping and sequencing analysis.
We genotyped the affected Ku10 proband and four of her unaffected siblings using Illumina Infinium HumanHap610W- Quad BeadChip genotyping arrays at the Australian Genome Research Facility (Melbourne, Australia). FEstim (5) was used to verify the reported consanguinity in this family.
We analyzed a subset of 11 711 SNP markers with high heterozygosity in approximate linkage equilibrium (one SNP was chosen per 0.3 cM) (12). Parametric multipoint linkage analysis was performed using MERLIN (13). We specified a fully penetrant recessive inheritance model with a 0% phenocopy rate, a disease allele frequency of 0.0001, and allele frequencies from the Centre d'Etude du Polymorphisme Humain (CEPH; Utah residents with ancestry from northern and western Europe) HapMap population.
Exome sequencing, alignment, variant detection and annotation
To efficiently identify a potential causative mutation, we sequenced the exomes of the Ku10 proband II-1 and her unaffected mother I-2. We additionally sequenced the exomes of the two affected children from Ku4, II-2 and II-3, along with the exome of their unaffected father I-1. Exome capture and sequencing was performed by Axeq Technologies (Rockville, MD, USA). DNA samples were enriched for ∼62 Mb of the genome using Illumina TruSeq capture, and sequenced using the Illumina HiSeq platform, generating 110 bp paired-end reads.
Reads were aligned to the hg19 reference genome using Novoalign version 2.07.09 (www.novocraft.com), with quality score recalibration performed. Multi-mapping reads were removed; potential PCR duplicates were discarded by using Picard version 1.21 (http://picard.sourceforge.net/). Supplementary Material, Table S1 summarizes the number of reads mapped for each exome and Supplementary Material, Table S2 summarizes the distribution of coverage for targeted bases.
Variants were called using the mpileup and bcftools view utilities from SAMtools version 0.1.18, (14,15) with low confidence variants discarded using the vcfutils.pl script from the same program. Variants were annotated using ANNOVAR (16). Variants were prioritized by filtering for variants located within linkage regions that were absent from or had an alternate allele frequency of ≤1% in public and in-house databases, were predicted to be protein-modifying, and had genotypes consistent with the hypothesized genetic model for each family. We used SIFT (17) and HumVar-trained PolyPhen-2 (18) (v2.2r398) to predict the biological impact of nonsynonymous variants.
Sanger sequencing
All exons, exon–intron boundaries and untranslated regions of CTSF were amplified by PCR. Primers were designed with the Primer 3 web application (http://frodo.wi.mit.edu/primer3). Sequences and PCR conditions are available upon request. PCR products were sequenced bi-directionally in triplicate with standard BigDye chemistry sequencing protocols. Electrophoresis of purified sequencing reactions was performed on an ABI 3730XL sequencing platform (Life Technologies Corporation, Carlsbad, CA, USA). Sequences were analyzed using the software program CodonCode.
Electron microscopy
Electron micrographs from cases with CTSF mutations, as well as from the animal studies were reviewed by one of us (S.C.).
In silico modeling of protein structure
Structural representations of human cathepsin F were generated using the RIBBONS program (19).
Ethics statement
Human studies were approved by the Human Research Ethics Committee of Austin Health, Melbourne, Australia. Informed consent was obtained from living subjects or their relatives. Some subjects had died as much as 20 years earlier, and autopsy material was collected and stored under the appropriate regulations of other participating hospitals or universities.
All experiments involving animal use were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Australian Government National Health and Medical Research Council (490037 to M.B., 628952 and 466671 to S.F.B., the Independent Research Institute Infrastructure Support Scheme to M.B. and K.R.S.); The Australian Research Council (FT100100764 to M.B.); the Victorian State Government (Operational Infrastructure Program to M.B. and K.R.S.); the Deutsche Forschungsgemeinschaft (SFB877 to P.S., J.G. and M.S.); the National Institutes of Health (NS41930 to J.D.C.); the Pratt Foundation (to K.R.S.), The Batten Disease Support and Research Association (to S.E.M., J.D.C., J.M.S. and K.B.S.) and the Natalie Fund (to J.D.C.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs Danielle Andrade, Umberto Aguglia, Stefano Meletti, Alessandro Simonati and Jay Pathmanathan for referral of cases.
Conflict of Interest statement. None declared.
References
- 1.Arsov T., Smith K.R., Damiano J., Franceschetti S., Canafoglia L., Bromhead C.J., Andermann E., Vears D.F., Cossette P., Rajagopalan S., et al. Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6. Am. J. Hum. Genet. 2011;88:566–573. doi: 10.1016/j.ajhg.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benitez B.A., Alvarado D., Cai Y., Mayo K., Chakraverty S., Norton J., Morris J.C., Sands M.S., Goate A., Cruchaga C. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS ONE. 2011;6:e26741. doi: 10.1371/journal.pone.0026741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nosková L., Stránecký V., Hartmannová H., Přistoupilová A., Barešová V., Ivánek R., Hůlková H., Jahnová H., Zee J.v.d., Staropoli J.F., et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011;89:241–252. doi: 10.1016/j.ajhg.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Velinov M., Dolzhanskaya N., Gonzalez M., Powell E., Konidari I., Hulme W., Staropoli J.F., Xin W., Wen G.Y., Barone R., et al. Mutations in the Gene DNAJC5 cause autosomal dominant Kufs disease in a proportion of cases: study of the Parry family and 8 other families. PLoS ONE. 2012;7:e29729. doi: 10.1371/journal.pone.0029729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leutenegger A.L., Prum B., Genin E., Verny C., Lemainque A., Clerget-Darpoux F., Thompson E.A., Leutenegger A.-L., Prum B., Genin E., et al. Estimation of the inbreeding coefficient through use of genomic data. Am. J. Hum. Genet. 2003;73:516–523. doi: 10.1086/378207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durbin R.M., Abecasis G.R., Altshuler D.L., Auton A., Brooks L.D., Gibbs R.A., Hurles M.E., McVean G.A. 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Somoza J.R., Palmer J.T., Ho J.D. The crystal structure of human cathepsin F and its implications for the development of novel immunomodulators. J. Mol. Biol. 2002;322:559–568. doi: 10.1016/s0022-2836(02)00780-5. [DOI] [PubMed] [Google Scholar]
- 8.Tang C.-H., Lee J.-W., Galvez M.G., Robillard L., Mole S.E., Chapman H.A. Murine cathepsin F deficiency causes neuronal lipofuscinosis and late-onset neurological disease. Mol. Cell. Biol. 2006;26:2309–2316. doi: 10.1128/MCB.26.6.2309-2316.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siintola E., Partanen S., Strömme P., Haapanen A., Haltia M., Maehlen J., Lehesjoki A.-E., Tyynelä J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–1445. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- 10.Dawson G., Dawson S.A., Siakotos A.N. Phospholipases and the molecular basis for the formation of ceroid in Batten disease. Adv. Exp. Med. Biol. 1989;266:259. doi: 10.1007/978-1-4899-5339-1_18. [DOI] [PubMed] [Google Scholar]
- 11.Shimmoto M., Takano T., Fukuhara Y., Oshima A., Sakuraba H., Suzuki Y. Japanese-type adult galactosialidosis: a unique and common splice junction mutation causing exon skipping in the protective protein/carboxypeptidase gene. Proc. Japan Acad., Ser. B Phys. Biol. Sci. 1990;66:217–222. [Google Scholar]
- 12.Bahlo M., Bromhead C.J. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009;25:1961–1962. doi: 10.1093/bioinformatics/btp313. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees.[see comment] Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 14.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar P., Henikoff S., Ng P.C., Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 18.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R., Adzhubei I.A., Schmidt S., et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraulis P.J. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 1991;24:946–950. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.