

Axonal transport defects in neurodegenerative diseases
- PMID: 19828789
- PMCID: PMC2801051
- DOI: 10.1523/JNEUROSCI.3463-09.2009
Axonal transport defects in neurodegenerative diseases
Abstract
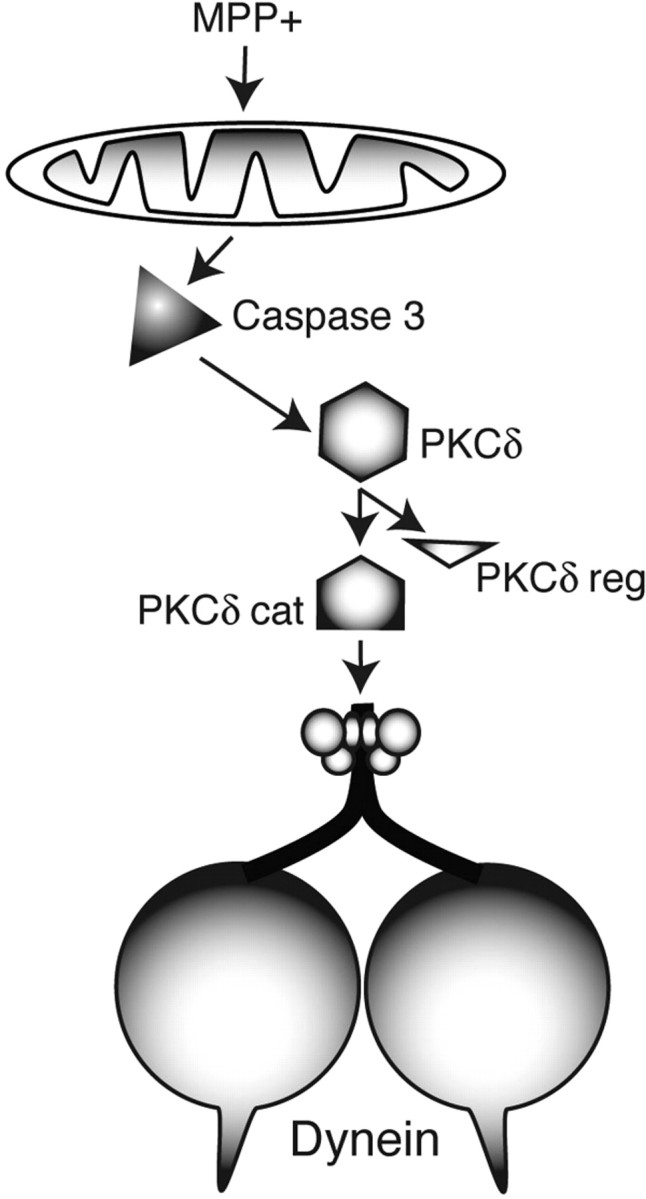
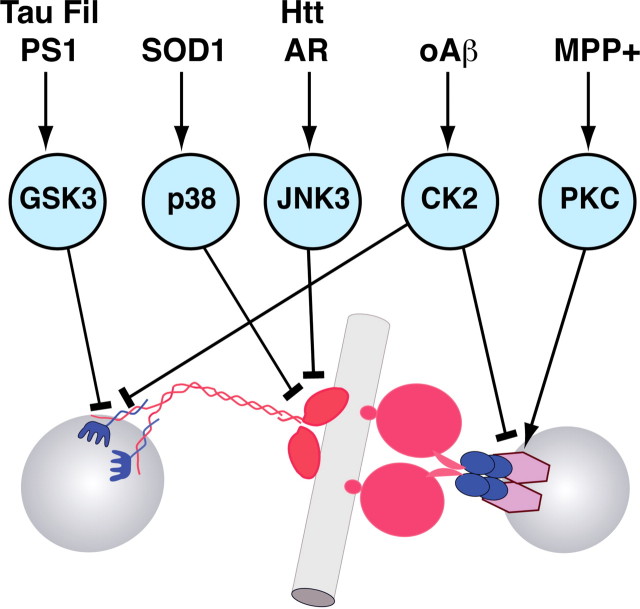
Adult-onset neurodegenerative diseases (AONDs) comprise a heterogeneous group of neurological disorders characterized by a progressive, age-dependent decline in neuronal function and loss of selected neuronal populations. Alterations in synaptic function and axonal connectivity represent early and critical pathogenic events in AONDs, but molecular mechanisms underlying these defects remain elusive. The large size and complex subcellular architecture of neurons render them uniquely vulnerable to alterations in axonal transport (AT). Accordingly, deficits in AT have been documented in most AONDs, suggesting a common defect acquired through different pathogenic pathways. These observations suggest that many AONDs can be categorized as dysferopathies, diseases in which alterations in AT represent a critical component in pathogenesis. Topics here address various molecular mechanisms underlying alterations in AT in several AONDs. Illumination of such mechanisms provides a framework for the development of novel therapeutic strategies aimed to prevent axonal and synaptic dysfunction in several major AONDs.
Figures
Similar articles
-
Regulation of motor proteins, axonal transport deficits and adult-onset neurodegenerative diseases.Neurobiol Dis. 2017 Sep;105:273-282. doi: 10.1016/j.nbd.2017.04.010. Epub 2017 Apr 11. Neurobiol Dis. 2017. PMID: 28411118 Free PMC article. Review.
-
α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies.Proc Natl Acad Sci U S A. 2018 Jul 24;115(30):7813-7818. doi: 10.1073/pnas.1713129115. Epub 2018 Jul 10. Proc Natl Acad Sci U S A. 2018. PMID: 29991596 Free PMC article.
-
Axonal degeneration in Alzheimer's disease: when signaling abnormalities meet the axonal transport system.Exp Neurol. 2013 Aug;246:44-53. doi: 10.1016/j.expneurol.2012.06.003. Epub 2012 Jun 19. Exp Neurol. 2013. PMID: 22721767 Free PMC article. Review.
-
Tau Isoforms Imbalance Impairs the Axonal Transport of the Amyloid Precursor Protein in Human Neurons.J Neurosci. 2017 Jan 4;37(1):58-69. doi: 10.1523/JNEUROSCI.2305-16.2016. J Neurosci. 2017. PMID: 28053030 Free PMC article.
-
Convergence of presenilin- and tau-mediated pathways on axonal trafficking and neuronal function.J Neurosci. 2010 Oct 6;30(40):13409-18. doi: 10.1523/JNEUROSCI.1964-10.2010. J Neurosci. 2010. PMID: 20926667 Free PMC article.
Cited by
-
Internalization and axonal transport of the HIV glycoprotein gp120.ASN Neuro. 2015 Jan 30;7(1):1759091414568186. doi: 10.1177/1759091414568186. Print 2015 Jan-Feb. ASN Neuro. 2015. PMID: 25636314 Free PMC article.
-
GSK3β is involved in the relief of mitochondria pausing in a Tau-dependent manner.PLoS One. 2011;6(11):e27686. doi: 10.1371/journal.pone.0027686. Epub 2011 Nov 14. PLoS One. 2011. PMID: 22110721 Free PMC article.
-
Reflectance speckle of retinal nerve fiber layer reveals axonal activity.Invest Ophthalmol Vis Sci. 2013 Apr 12;54(4):2616-23. doi: 10.1167/iovs.12-11347. Invest Ophthalmol Vis Sci. 2013. PMID: 23532525 Free PMC article.
-
Neurodegeneration as an RNA disorder.Prog Neurobiol. 2012 Dec;99(3):293-315. doi: 10.1016/j.pneurobio.2012.09.006. Epub 2012 Oct 10. Prog Neurobiol. 2012. PMID: 23063563 Free PMC article. Review.
-
Drug treatment for chemotherapy-induced peripheral neuropathy in patients with pancreatic cancer.Fukushima J Med Sci. 2022 Apr 8;68(1):1-10. doi: 10.5387/fms.2021-32. Epub 2022 Feb 23. Fukushima J Med Sci. 2022. PMID: 35197393 Free PMC article. Review.
References
-
- Ackerley S, Grierson AJ, Banner S, Perkinton MS, Brownlees J, Byers HL, Ward M, Thornhill P, Hussain K, Waby JS, Anderton BH, Cooper JD, Dingwall C, Leigh PN, Shaw CE, Miller CC. p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol Cell Neurosci. 2004;26:354–364. - PubMed
-
- Albin RL, Reiner A, Anderson KD, Dure LS, 4th, Handelin B, Balfour R, Whetsell WO, Jr, Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31:425–430. - PubMed
-
- Andersen JK, Kumar J, Srinivas B, Kaur D, Hsu M, Rajagopalan S. The hunt for a cure for Parkinson's disease. Sci Aging Knowledge Environ. 2001;2001:re1. - PubMed
-
- Baas PW, Karabay A, Qiang L. Microtubules cut and run. Trends Cell Biol. 2005;15:518–524. - PubMed
-
- Bendotti C, Atzori C, Piva R, Tortarolo M, Strong MJ, DeBiasi S, Migheli A. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J Neuropathol Exp Neurol. 2004;63:113–119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical