

Neuroprotection and lifespan extension in Ppt1(-/-) mice by NtBuHA: therapeutic implications for INCL
- PMID: 24056696
- PMCID: PMC3812271
- DOI: 10.1038/nn.3526
Neuroprotection and lifespan extension in Ppt1(-/-) mice by NtBuHA: therapeutic implications for INCL
Abstract
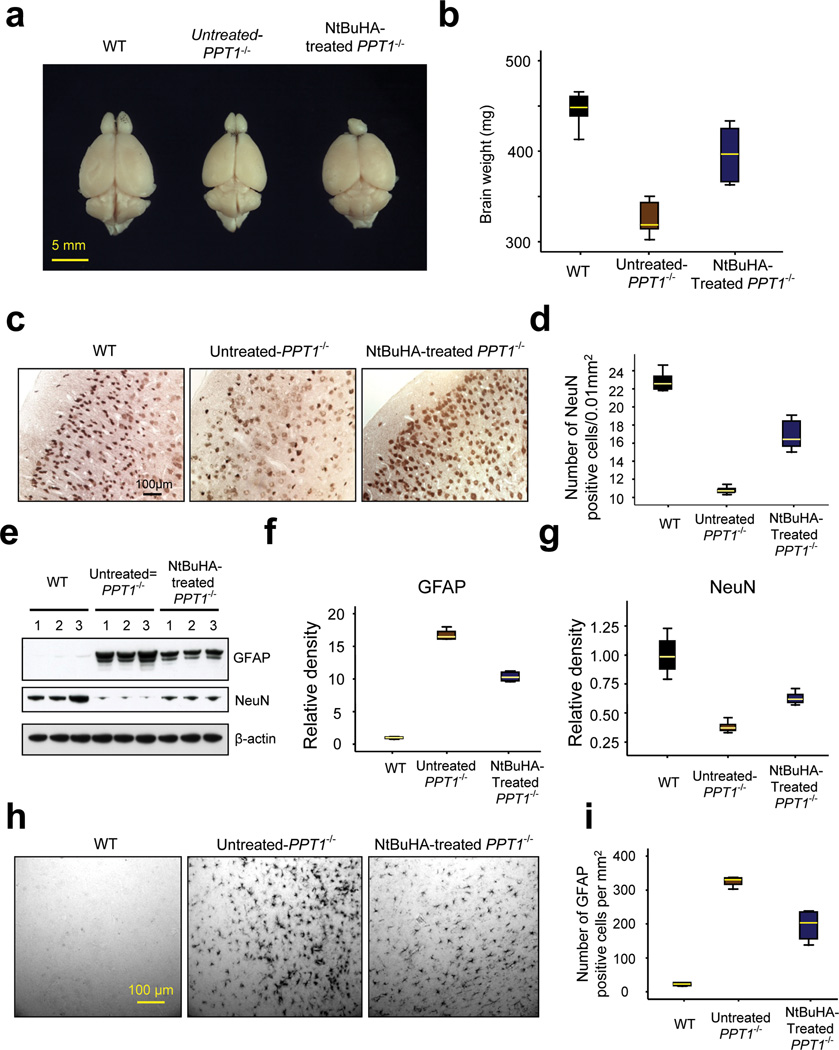
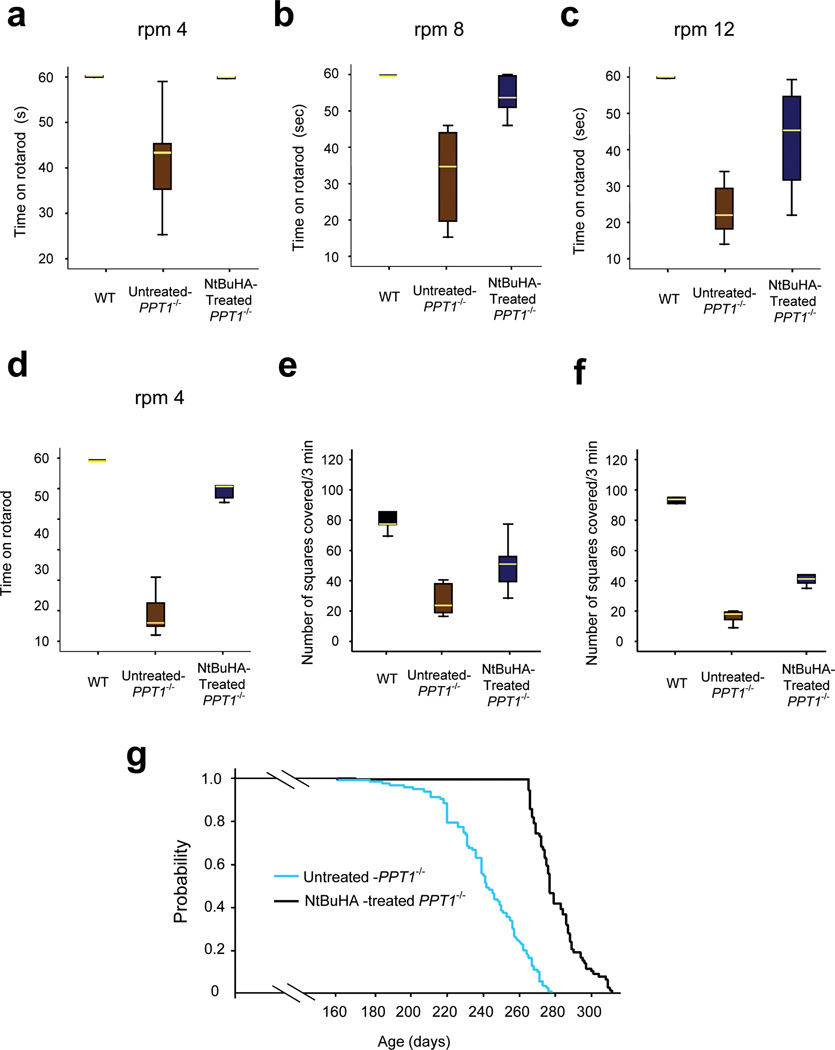
Infantile neuronal ceroid lipofuscinosis (INCL) is a devastating childhood neurodegenerative lysosomal storage disease (LSD) that has no effective treatment. It is caused by inactivating mutations in the palmitoyl-protein thioesterase-1 (PPT1) gene. PPT1 deficiency impairs the cleavage of thioester linkage in palmitoylated proteins (constituents of ceroid), preventing degradation by lysosomal hydrolases. Consequently, accumulation of lysosomal ceroid leads to INCL. Thioester linkage is cleaved by nucleophilic attack. Hydroxylamine, a potent nucleophilic cellular metabolite, may have therapeutic potential for INCL, but its toxicity precludes clinical application. We found that a hydroxylamine derivative, N-(tert-Butyl) hydroxylamine (NtBuHA), was non-toxic, cleaved thioester linkage in palmitoylated proteins and mediated lysosomal ceroid depletion in cultured cells from INCL patients. In Ppt1(-/-) mice, which mimic INCL, NtBuHA crossed the blood-brain barrier, depleted lysosomal ceroid, suppressed neuronal apoptosis, slowed neurological deterioration and extended lifespan. Our findings provide a proof of concept that thioesterase-mimetic and antioxidant small molecules such as NtBuHA are potential drug targets for thioesterase deficiency diseases such as INCL.
Figures
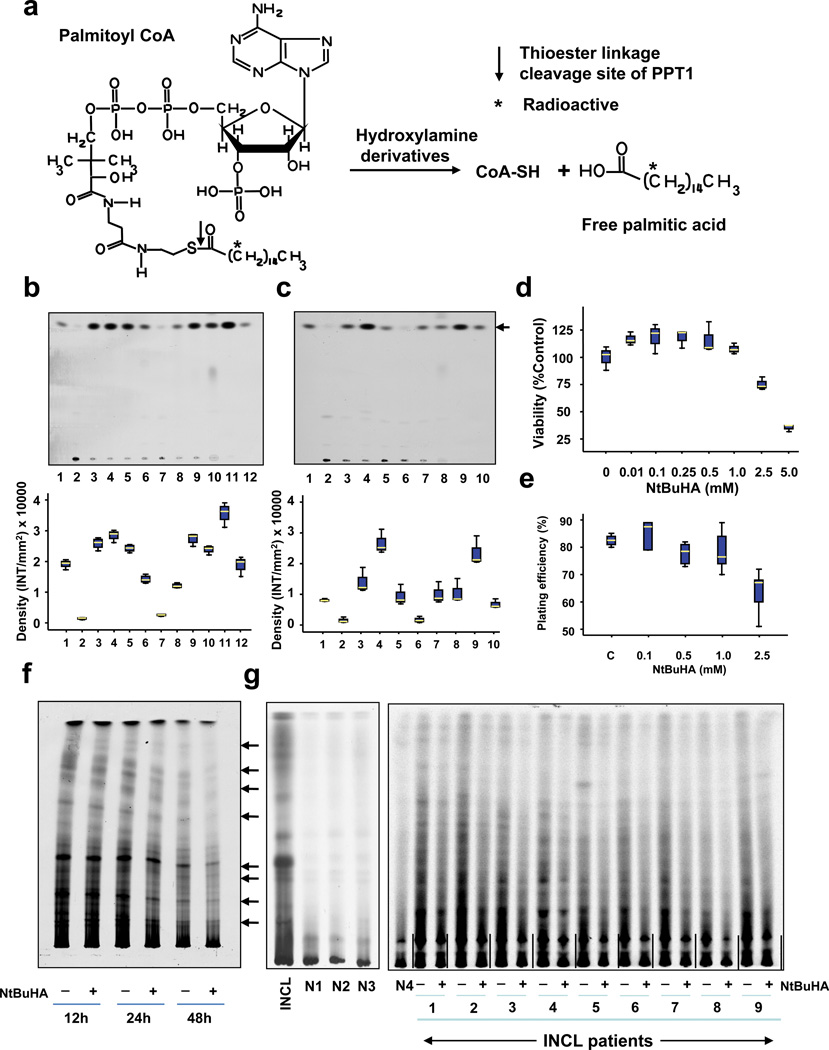
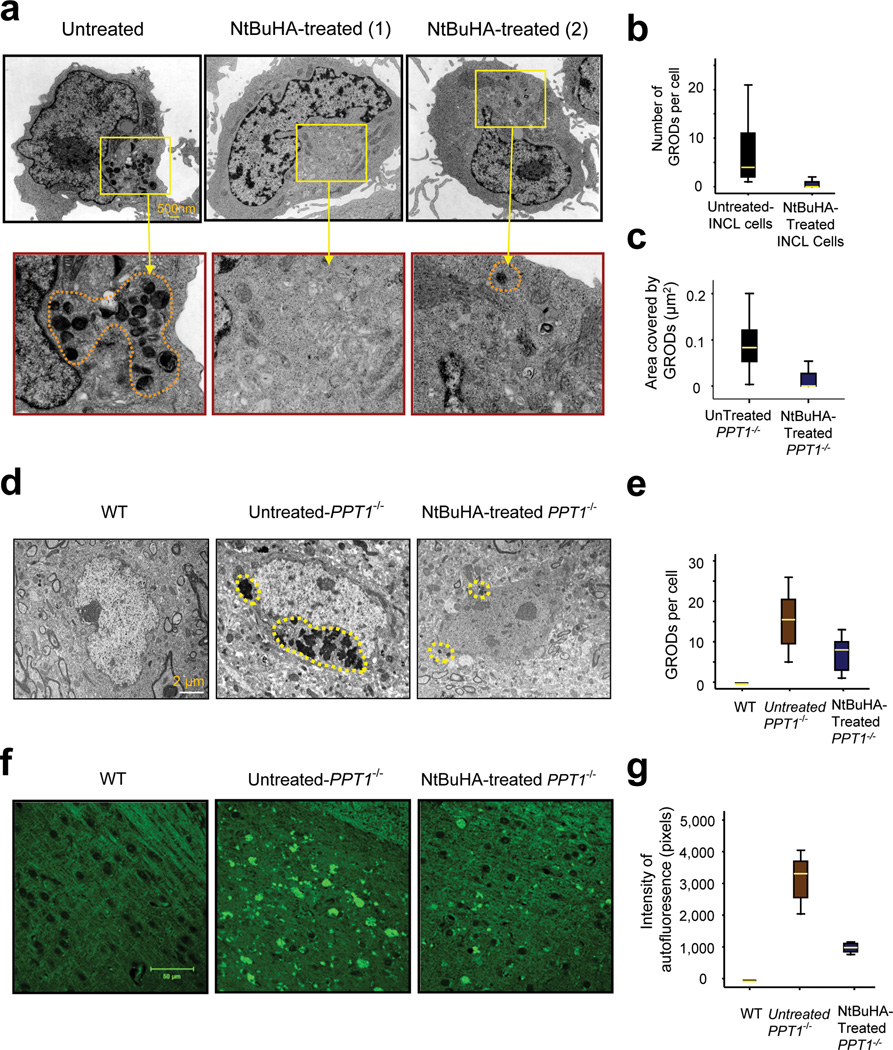
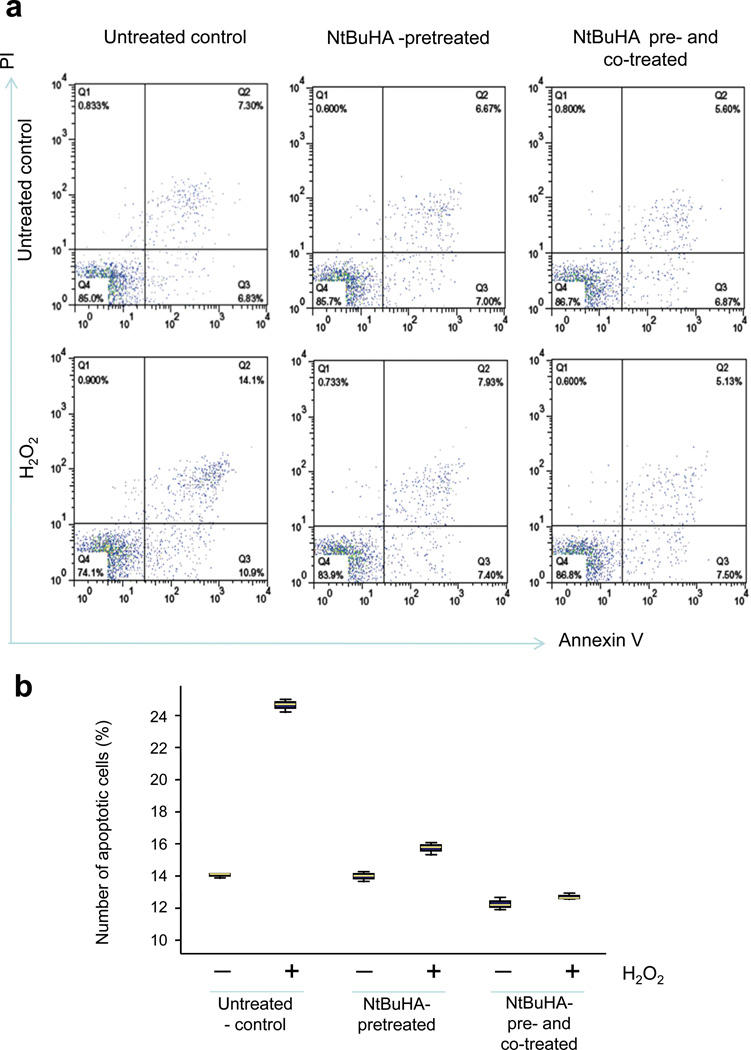
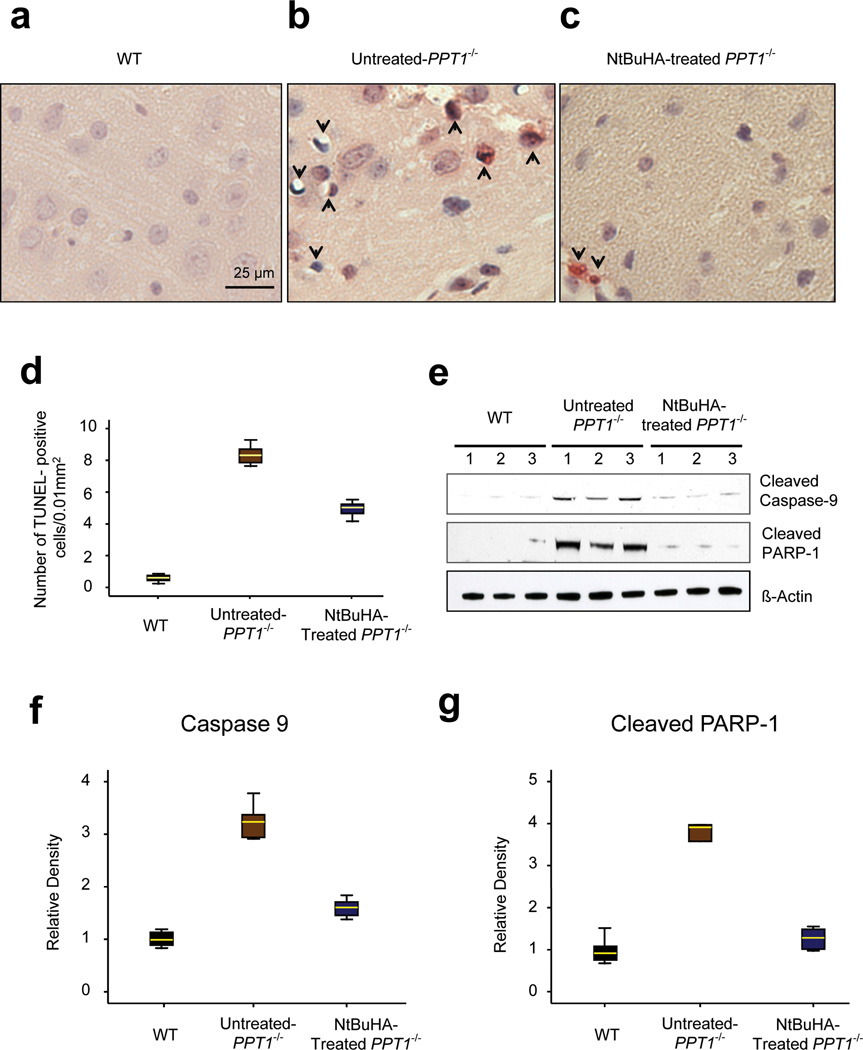
Comment in
-
Lysosomal storage diseases: Thioesterase mimetic reduces toxicity.Nat Rev Drug Discov. 2013 Nov;12(11):828. doi: 10.1038/nrd4156. Epub 2013 Oct 18. Nat Rev Drug Discov. 2013. PMID: 24136395 No abstract available.
Similar articles
-
Cln1-mutations suppress Rab7-RILP interaction and impair autophagy contributing to neuropathology in a mouse model of infantile neuronal ceroid lipofuscinosis.J Inherit Metab Dis. 2020 Sep;43(5):1082-1101. doi: 10.1002/jimd.12242. Epub 2020 Apr 27. J Inherit Metab Dis. 2020. PMID: 32279353 Free PMC article.
-
Cln1 gene disruption in mice reveals a common pathogenic link between two of the most lethal childhood neurodegenerative lysosomal storage disorders.Hum Mol Genet. 2015 Oct 1;24(19):5416-32. doi: 10.1093/hmg/ddv266. Epub 2015 Jul 9. Hum Mol Genet. 2015. PMID: 26160911 Free PMC article.
-
Reduction of neuroinflammation and seizures in a mouse model of CLN1 batten disease using the small molecule enzyme mimetic, N-Tert-butyl hydroxylamine.Mol Genet Metab. 2024 Sep-Oct;143(1-2):108537. doi: 10.1016/j.ymgme.2024.108537. Epub 2024 Jul 15. Mol Genet Metab. 2024. PMID: 39033629
-
Pathogenesis and therapies for infantile neuronal ceroid lipofuscinosis (infantile CLN1 disease).Biochim Biophys Acta. 2013 Nov;1832(11):1906-9. doi: 10.1016/j.bbadis.2013.05.026. Epub 2013 Jun 6. Biochim Biophys Acta. 2013. PMID: 23747979 Free PMC article. Review.
-
Protein acyl thioesterases (Review).Mol Membr Biol. 2009 Jan;26(1):32-41. doi: 10.1080/09687680802629329. Epub 2008 Dec 29. Mol Membr Biol. 2009. PMID: 19115143 Review.
Cited by
-
APT1-Mediated Depalmitoylation Regulates Hippocampal Synaptic Plasticity.J Neurosci. 2022 Mar 30;42(13):2662-2677. doi: 10.1523/JNEUROSCI.1741-21.2022. Epub 2022 Feb 14. J Neurosci. 2022. PMID: 35165175 Free PMC article.
-
Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses.CNS Drugs. 2019 Apr;33(4):315-325. doi: 10.1007/s40263-019-00620-8. CNS Drugs. 2019. PMID: 30877620 Free PMC article. Review.
-
Non-invasive assessment of retinal alterations in mouse models of infantile and juvenile neuronal ceroid lipofuscinosis by spectral domain optical coherence tomography.Acta Neuropathol Commun. 2014 May 10;2:54. doi: 10.1186/2051-5960-2-54. Acta Neuropathol Commun. 2014. PMID: 24887158 Free PMC article.
-
Progress in the Development of Small Molecule Therapeutics for the Treatment of Neuronal Ceroid Lipofuscinoses (NCLs).J Med Chem. 2016 May 26;59(10):4415-27. doi: 10.1021/acs.jmedchem.5b01020. Epub 2015 Nov 24. J Med Chem. 2016. PMID: 26565590 Free PMC article. Review.
-
S-Palmitoylation of Synaptic Proteins in Neuronal Plasticity in Normal and Pathological Brains.Cells. 2023 Jan 21;12(3):387. doi: 10.3390/cells12030387. Cells. 2023. PMID: 36766729 Free PMC article. Review.
References
-
- Cox TM, Cachón-González MB. The cellular pathology of lysosomal diseases. J. Pathol. 2012;226:241–254. - PubMed
-
- Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat. Rev. Neurosci. 2005;6:713–725. - PubMed
-
- Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim. Biophys. Acta. 2013;1832:1807–1826. - PubMed
-
- Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum.Mutat. 2012;33:42–63. - PubMed
-
- Haltia M, Goebel HH. The neuronal ceroid lipofuscinoses: A historical introduction. Biochim Biophys Acta. 2013;1832:1795–1800. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
