

Sodium channel dysfunction in intractable childhood epilepsy with generalized tonic-clonic seizures
- PMID: 16210358
- PMCID: PMC1464244
- DOI: 10.1113/jphysiol.2005.094326
Sodium channel dysfunction in intractable childhood epilepsy with generalized tonic-clonic seizures
Abstract
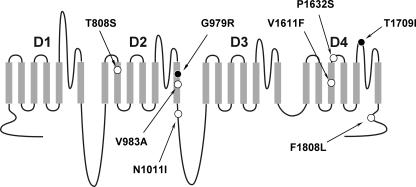
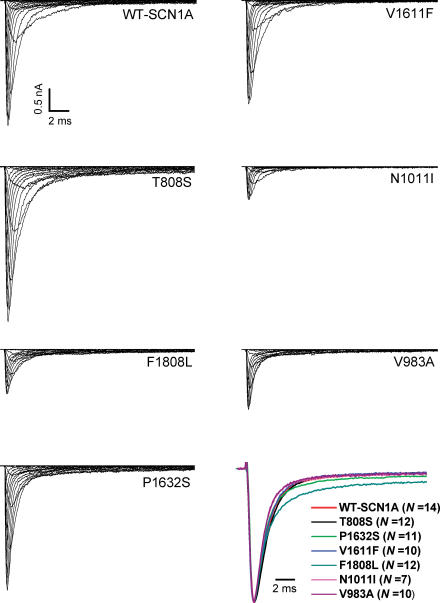
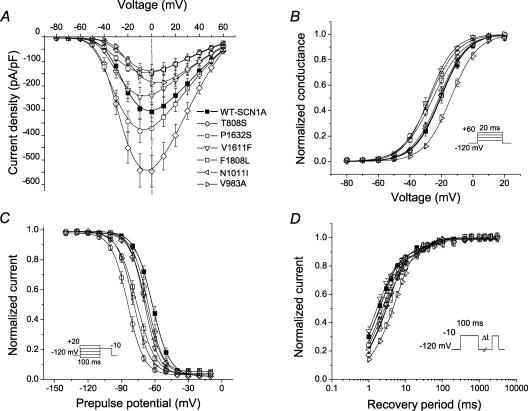
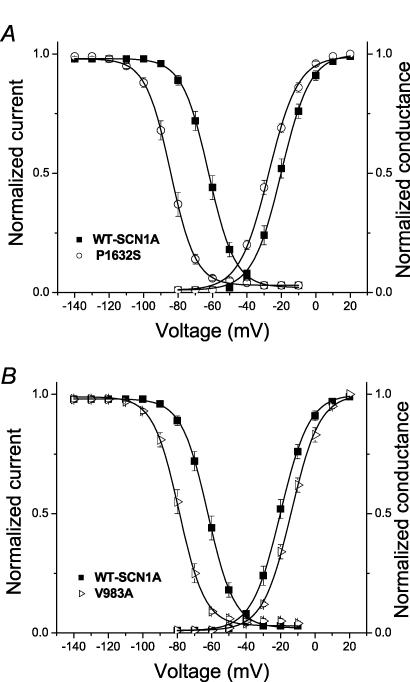
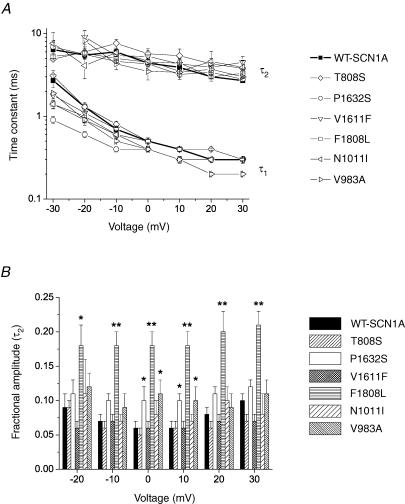
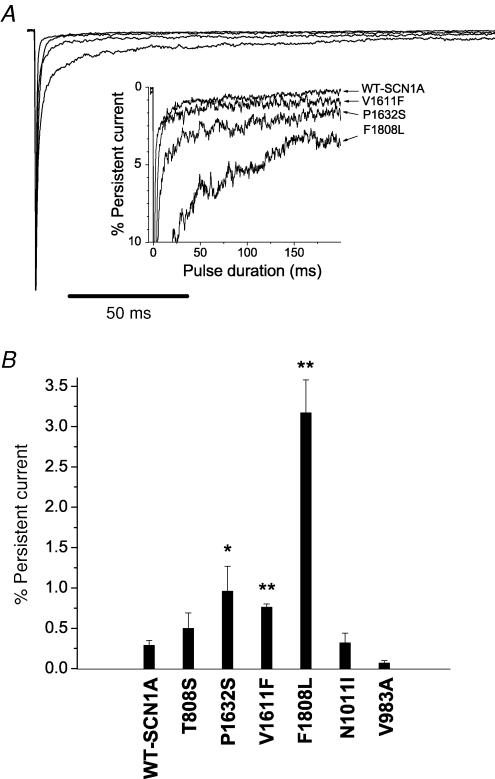
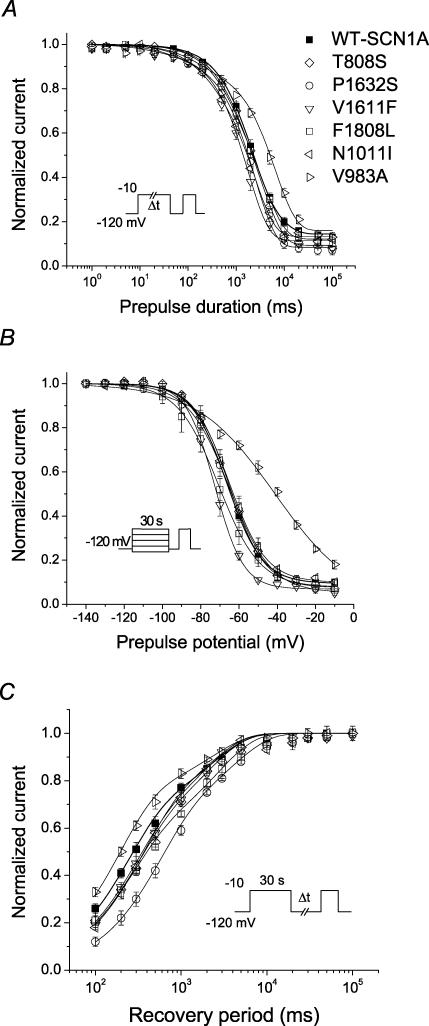
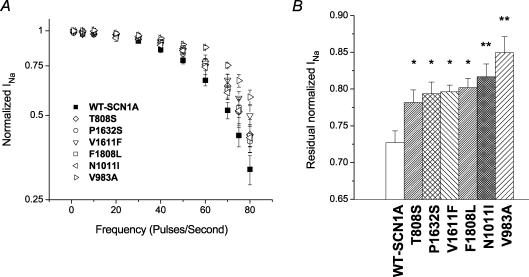
Mutations in SCN1A, the gene encoding the brain voltage-gated sodium channel alpha(1) subunit (Na(V)1.1), are associated with genetic forms of epilepsy, including generalized epilepsy with febrile seizures plus (GEFS+ type 2), severe myoclonic epilepsy of infancy (SMEI) and related conditions. Several missense SCN1A mutations have been identified in probands affected by the syndrome of intractable childhood epilepsy with generalized tonic-clonic seizures (ICEGTC), which bears similarity to SMEI. To test whether ICEGTC arises from molecular mechanisms similar to those involved in SMEI, we characterized eight ICEGTC missense mutations by whole-cell patch clamp recording of recombinant human SCN1A heterologously expressed in cultured mammalian cells. Two mutations (G979R and T1709I) were non-functional. The remaining alleles (T808S, V983A, N1011I, V1611F, P1632S and F1808L) exhibited measurable sodium current, but had heterogeneous biophysical phenotypes. Mutant channels exhibited lower (V983A, N1011I and F1808L), greater (T808S) or similar (V1611F and P1632S) peak sodium current densities compared with wild-type (WT) SCN1A. Three mutations (V1611F, P1632S and F1808L) displayed hyperpolarized conductance-voltage relationships, while V983A exhibited a strong depolarizing shift in the voltage dependence of activation. All mutants except T808S had hyperpolarized shifts in the voltage dependence of steady-state channel availability. Three mutants (V1611F, P1632S and F1808L) exhibited persistent sodium current ranging from approximately 1-3% of peak current amplitude that was significantly greater than WT-SCN1A. Several mutants had impaired slow inactivation, with V983A showing the most prominent effect. Finally, all of the functional alleles exhibited reduced use-dependent channel inhibition. In summary, SCN1A mutations associated with ICEGTC result in a wide spectrum of biophysical defects, including mild-to-moderate gating impairments, shifted voltage dependence and reduced use dependence. The constellation of biophysical abnormalities for some mutants is distinct from those previously observed for GEFS+ and SMEI, suggesting possible, but complex, genotype-phenotype correlations.
Figures
Comment in
-
(What to do) when epilepsy gene mutations stop making sense.Epilepsy Curr. 2007 Jan-Feb;7(1):23-5. doi: 10.1111/j.1535-7511.2007.00157.x. Epilepsy Curr. 2007. PMID: 17304347 Free PMC article. No abstract available.
Similar articles
-
Single-channel properties of human NaV1.1 and mechanism of channel dysfunction in SCN1A-associated epilepsy.J Gen Physiol. 2006 Jan;127(1):1-14. doi: 10.1085/jgp.200509373. J Gen Physiol. 2006. PMID: 16380441 Free PMC article.
-
Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A.J Neurosci. 2003 Dec 10;23(36):11289-95. doi: 10.1523/JNEUROSCI.23-36-11289.2003. J Neurosci. 2003. PMID: 14672992 Free PMC article.
-
Nonfunctional SCN1A is common in severe myoclonic epilepsy of infancy.Epilepsia. 2006 Oct;47(10):1636-42. doi: 10.1111/j.1528-1167.2006.00643.x. Epilepsia. 2006. PMID: 17054685
-
Clinical spectrum of mutations in SCN1A gene: severe myoclonic epilepsy in infancy and related epilepsies.Epilepsy Res. 2006 Aug;70 Suppl 1:S223-30. doi: 10.1016/j.eplepsyres.2006.01.019. Epub 2006 Jun 27. Epilepsy Res. 2006. PMID: 16806826 Review.
-
Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity.Neurology. 2004 Jul 27;63(2):329-34. doi: 10.1212/01.wnl.0000129829.31179.5b. Neurology. 2004. PMID: 15277629 Review.
Cited by
-
Persistent sodium current and its role in epilepsy.Epilepsy Curr. 2007 Jan-Feb;7(1):15-22. doi: 10.1111/j.1535-7511.2007.00156.x. Epilepsy Curr. 2007. PMID: 17304346 Free PMC article.
-
Gain of Function for the SCN1A/hNav1.1-L1670W Mutation Responsible for Familial Hemiplegic Migraine.Front Mol Neurosci. 2018 Jul 9;11:232. doi: 10.3389/fnmol.2018.00232. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30038559 Free PMC article.
-
Ionic mechanisms of endogenous bursting in CA3 hippocampal pyramidal neurons: a model study.PLoS One. 2008 Apr 30;3(4):e2056. doi: 10.1371/journal.pone.0002056. PLoS One. 2008. PMID: 18446231 Free PMC article.
-
Increased Persistent Sodium Current Causes Neuronal Hyperexcitability in the Entorhinal Cortex of Fmr1 Knockout Mice.Cell Rep. 2016 Sep 20;16(12):3157-3166. doi: 10.1016/j.celrep.2016.08.046. Cell Rep. 2016. PMID: 27653682 Free PMC article.
-
(What to do) when epilepsy gene mutations stop making sense.Epilepsy Curr. 2007 Jan-Feb;7(1):23-5. doi: 10.1111/j.1535-7511.2007.00157.x. Epilepsy Curr. 2007. PMID: 17304347 Free PMC article. No abstract available.
References
-
- Abou-Khalil B, Ge Q, Desai R, Ryther R, Bazyk A, Bailey R, et al. Partial epilepsy and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology. 2001;57:2265–2272. - PubMed
-
- Babitch JA, Anthony FA. Grasping for calcium binding sites in sodium channels with an EF hand. J Theor Biol. 1987;127:451–459. - PubMed
-
- Bennett PB, Yazawa K, Makita N, George AL., Jr Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. - PubMed
-
- Cannon SC, Strittmatter SM. Functional expression of sodium channel mutations identified in families with periodic paralysis. Neuron. 1993;10:317–326. - PubMed
-
- Claes L, Ceulemans B, Audenaert D, Smets K, Lofgren A, Del Favero J, et al. De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat. 2003;21:615–621. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases