

The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities
- PMID: 26166479
- PMCID: PMC4573249
- DOI: 10.1016/j.ajhg.2015.06.009
The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities
Abstract
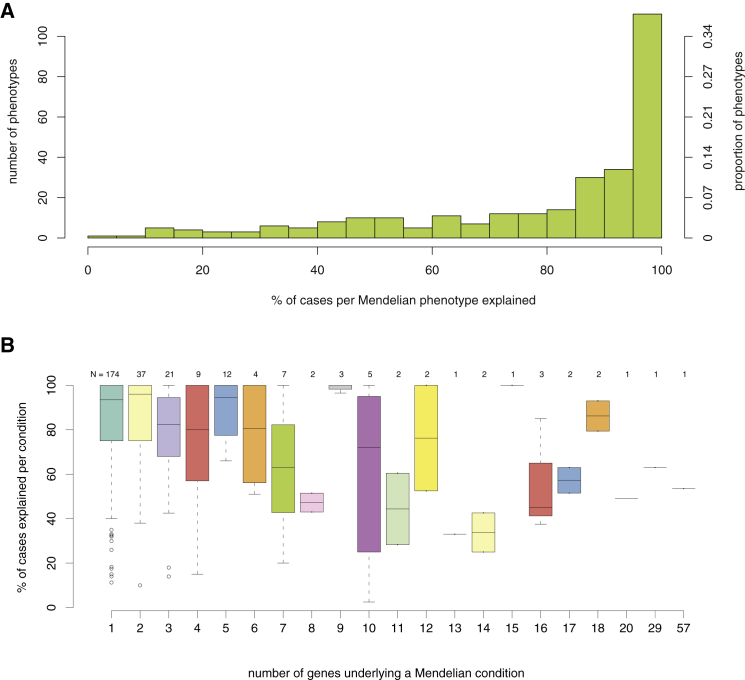
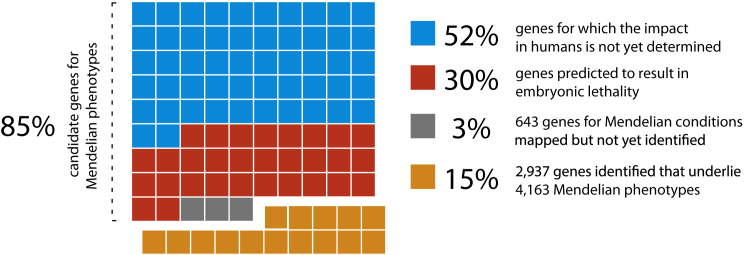
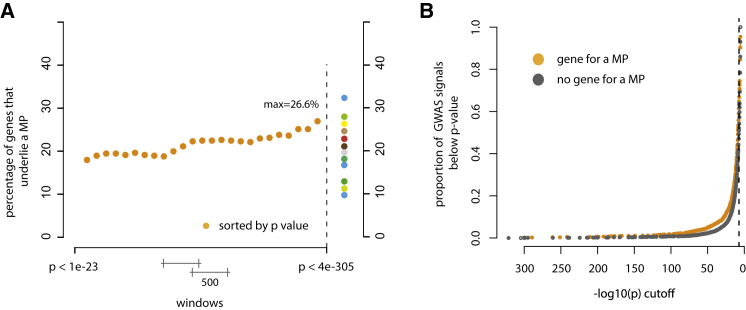
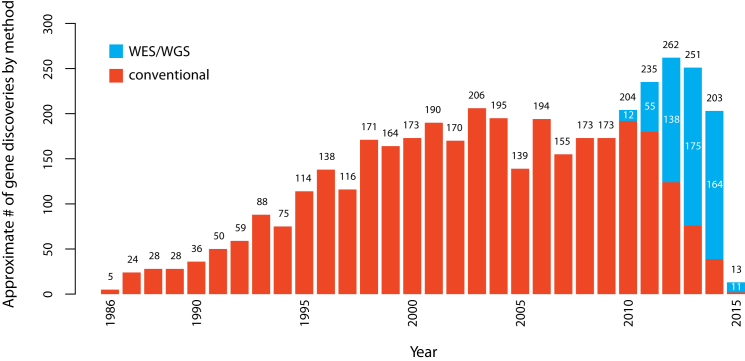
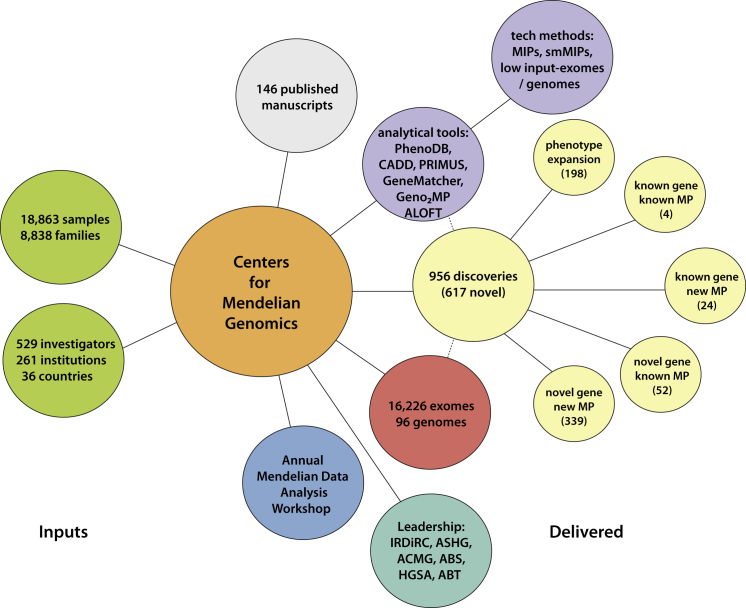
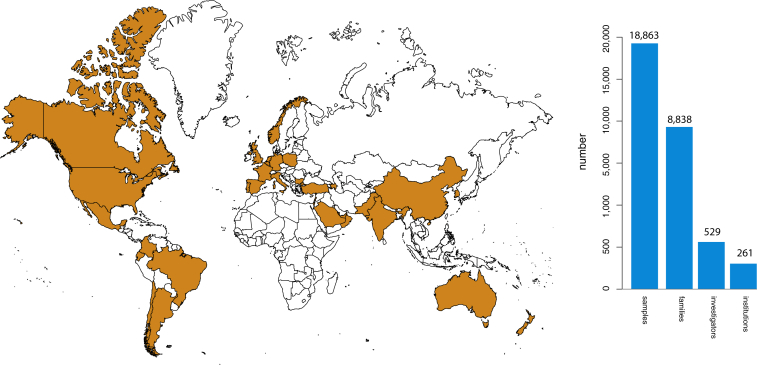
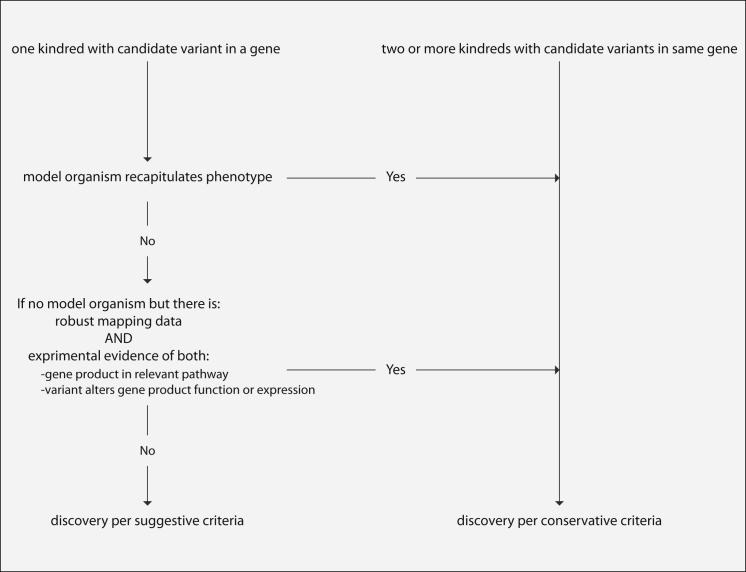
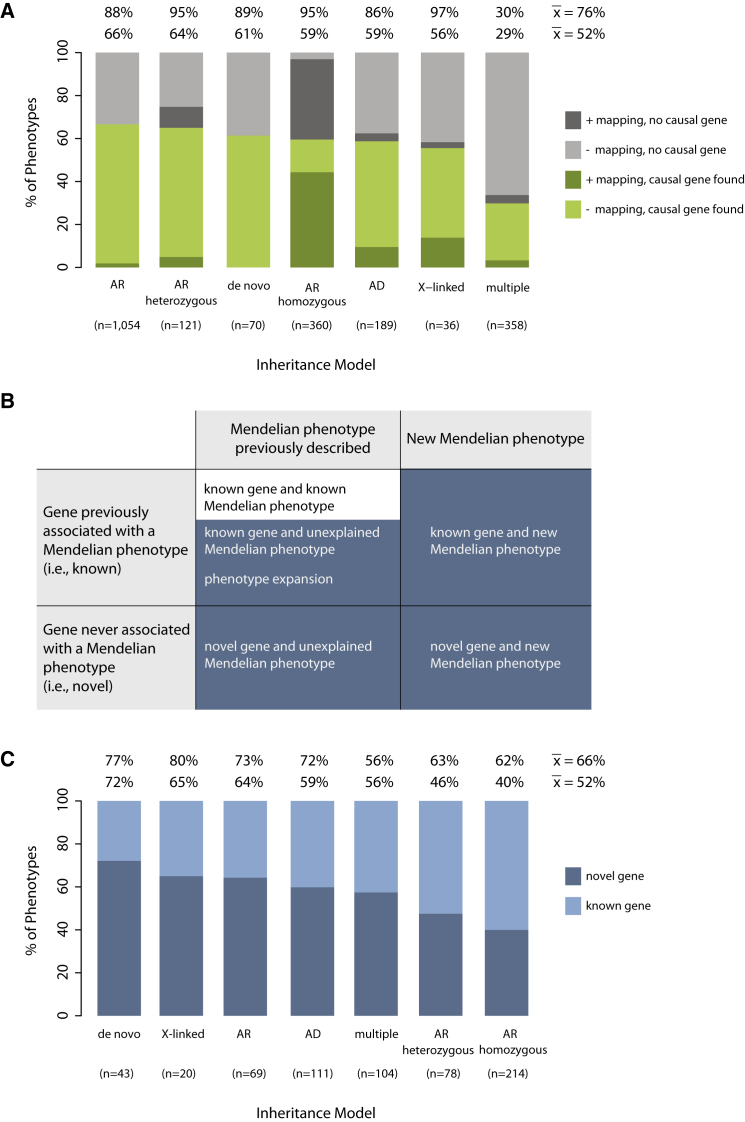
Discovering the genetic basis of a Mendelian phenotype establishes a causal link between genotype and phenotype, making possible carrier and population screening and direct diagnosis. Such discoveries also contribute to our knowledge of gene function, gene regulation, development, and biological mechanisms that can be used for developing new therapeutics. As of February 2015, 2,937 genes underlying 4,163 Mendelian phenotypes have been discovered, but the genes underlying ∼50% (i.e., 3,152) of all known Mendelian phenotypes are still unknown, and many more Mendelian conditions have yet to be recognized. This is a formidable gap in biomedical knowledge. Accordingly, in December 2011, the NIH established the Centers for Mendelian Genomics (CMGs) to provide the collaborative framework and infrastructure necessary for undertaking large-scale whole-exome sequencing and discovery of the genetic variants responsible for Mendelian phenotypes. In partnership with 529 investigators from 261 institutions in 36 countries, the CMGs assessed 18,863 samples from 8,838 families representing 579 known and 470 novel Mendelian phenotypes as of January 2015. This collaborative effort has identified 956 genes, including 375 not previously associated with human health, that underlie a Mendelian phenotype. These results provide insight into study design and analytical strategies, identify novel mechanisms of disease, and reveal the extensive clinical variability of Mendelian phenotypes. Discovering the gene underlying every Mendelian phenotype will require tackling challenges such as worldwide ascertainment and phenotypic characterization of families affected by Mendelian conditions, improvement in sequencing and analytical techniques, and pervasive sharing of phenotypic and genomic data among researchers, clinicians, and families.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
The Centers for Mendelian Genomics: a new large-scale initiative to identify the genes underlying rare Mendelian conditions.Am J Med Genet A. 2012 Jul;158A(7):1523-5. doi: 10.1002/ajmg.a.35470. Epub 2012 May 24. Am J Med Genet A. 2012. PMID: 22628075 Free PMC article.
-
Insights into genetics, human biology and disease gleaned from family based genomic studies.Genet Med. 2019 Apr;21(4):798-812. doi: 10.1038/s41436-018-0408-7. Epub 2019 Jan 18. Genet Med. 2019. PMID: 30655598 Free PMC article. Review.
-
Centers for Mendelian Genomics: A decade of facilitating gene discovery.Genet Med. 2022 Apr;24(4):784-797. doi: 10.1016/j.gim.2021.12.005. Epub 2022 Feb 9. Genet Med. 2022. PMID: 35148959 Free PMC article. Review.
-
Exome sequencing for gene discovery in lethal fetal disorders--harnessing the value of extreme phenotypes.Prenat Diagn. 2015 Oct;35(10):1005-9. doi: 10.1002/pd.4464. Epub 2014 Aug 22. Prenat Diagn. 2015. PMID: 25046514 Review.
-
A Genocentric Approach to Discovery of Mendelian Disorders.Am J Hum Genet. 2019 Nov 7;105(5):974-986. doi: 10.1016/j.ajhg.2019.09.027. Epub 2019 Oct 24. Am J Hum Genet. 2019. PMID: 31668702 Free PMC article.
Cited by
-
PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions.PLoS Comput Biol. 2016 May 25;12(5):e1004962. doi: 10.1371/journal.pcbi.1004962. eCollection 2016 May. PLoS Comput Biol. 2016. PMID: 27224906 Free PMC article.
-
The role of small in-frame insertions/deletions in inherited eye disorders and how structural modelling can help estimate their pathogenicity.Orphanet J Rare Dis. 2016 Sep 14;11(1):125. doi: 10.1186/s13023-016-0505-0. Orphanet J Rare Dis. 2016. PMID: 27628848 Free PMC article.
-
Personalized structural biology reveals the molecular mechanisms underlying heterogeneous epileptic phenotypes caused by de novo KCNC2 variants.HGG Adv. 2022 Jul 19;3(4):100131. doi: 10.1016/j.xhgg.2022.100131. eCollection 2022 Oct 13. HGG Adv. 2022. PMID: 36035247 Free PMC article.
-
Panomics: New Databases for Advancing Cardiology.Front Cardiovasc Med. 2021 May 10;8:587768. doi: 10.3389/fcvm.2021.587768. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 34041278 Free PMC article. Review.
-
Ultra-rare Disease and Genomics-Driven Precision Medicine.Genomics Inform. 2016 Jun;14(2):42-5. doi: 10.5808/GI.2016.14.2.42. Epub 2016 Jun 30. Genomics Inform. 2016. PMID: 27445646 Free PMC article. Review.
References
-
- Green E.D., Guyer M.S., National Human Genome Research Institute Charting a course for genomic medicine from base pairs to bedside. Nature. 2011;470:204–213. - PubMed
-
- Kaiser J. Human genetics. Affordable ‘exomes’ fill gaps in a catalog of rare diseases. Science. 2010;330:903. - PubMed
-
- Stankiewicz P., Lupski J.R. Structural variation in the human genome and its role in disease. Annu. Rev. Med. 2010;61:437–455. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
