

Long-read human genome sequencing and its applications
- PMID: 32504078
- PMCID: PMC7877196
- DOI: 10.1038/s41576-020-0236-x
Long-read human genome sequencing and its applications
Abstract
Over the past decade, long-read, single-molecule DNA sequencing technologies have emerged as powerful players in genomics. With the ability to generate reads tens to thousands of kilobases in length with an accuracy approaching that of short-read sequencing technologies, these platforms have proven their ability to resolve some of the most challenging regions of the human genome, detect previously inaccessible structural variants and generate some of the first telomere-to-telomere assemblies of whole chromosomes. Long-read sequencing technologies will soon permit the routine assembly of diploid genomes, which will revolutionize genomics by revealing the full spectrum of human genetic variation, resolving some of the missing heritability and leading to the discovery of novel mechanisms of disease.
Conflict of interest statement
COMPETING INTERESTS
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc.
Figures
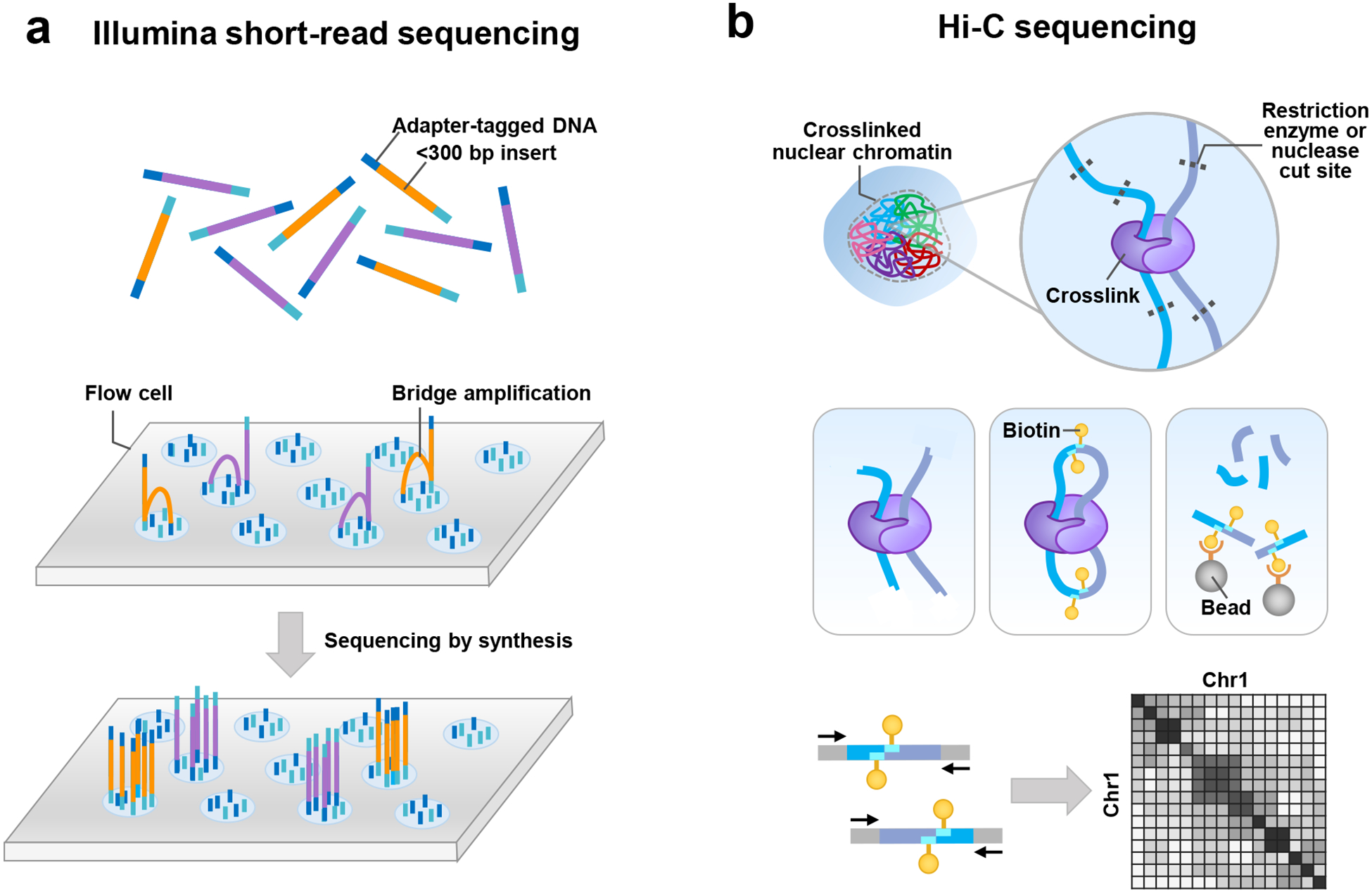
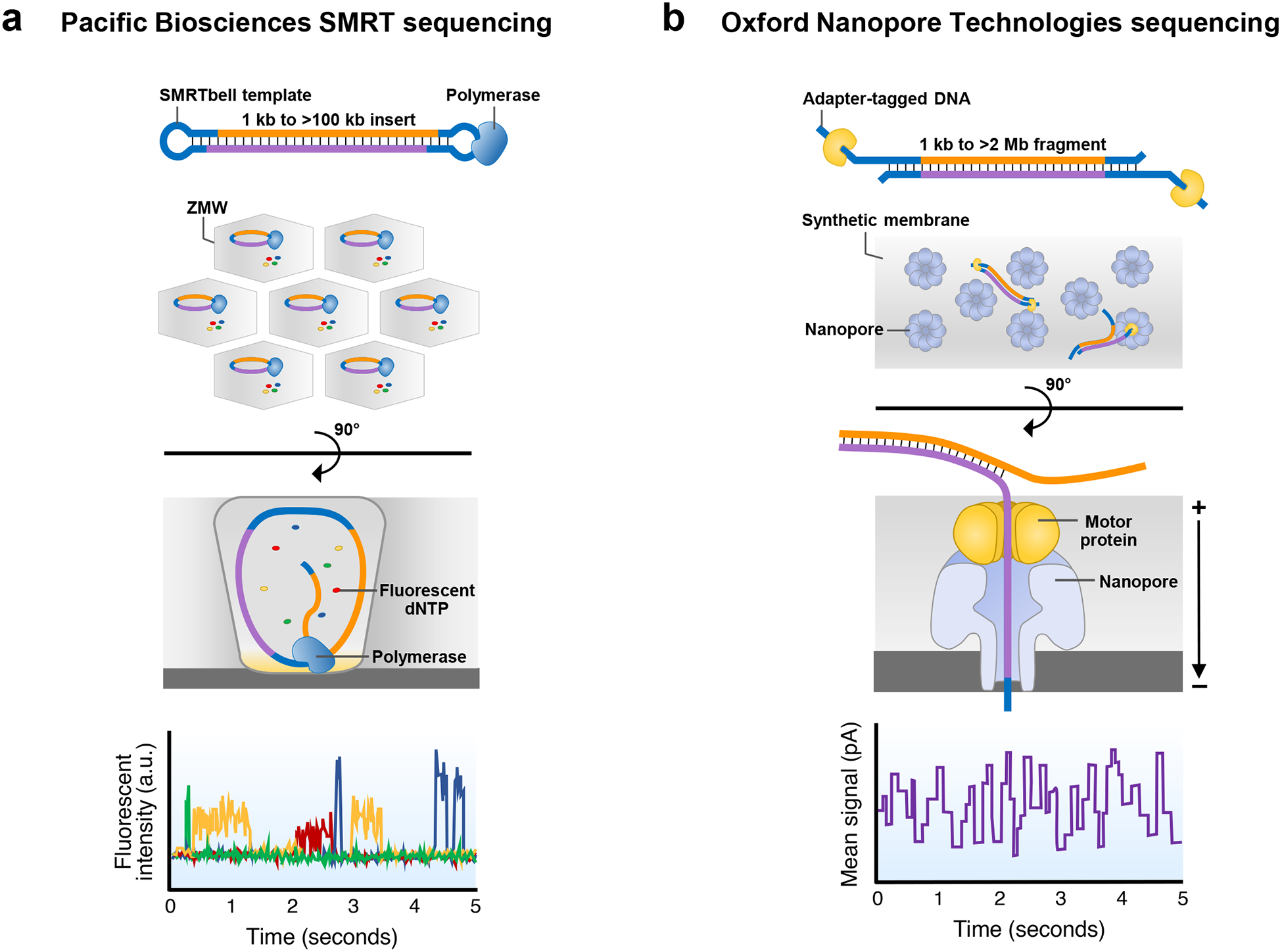
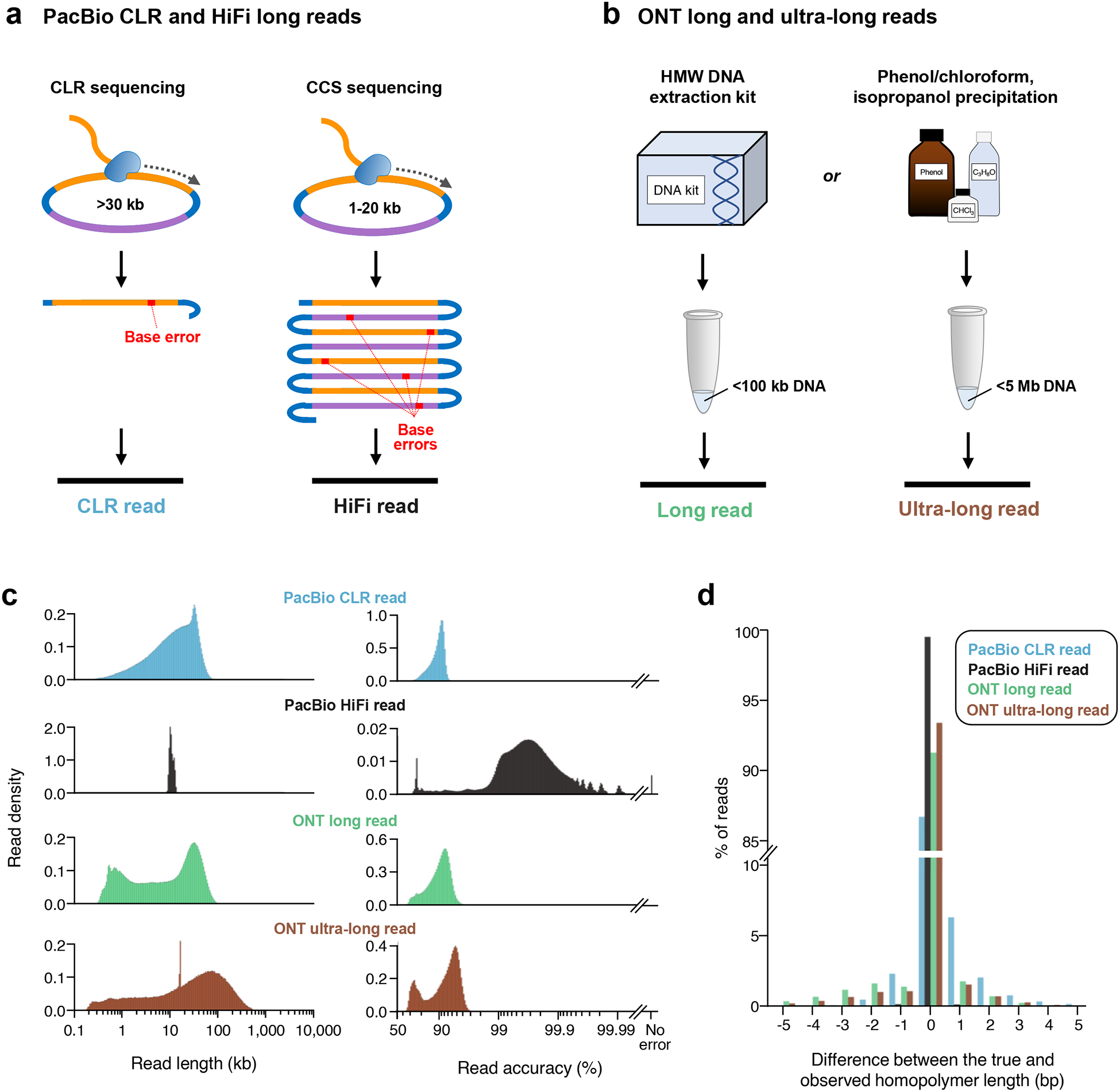
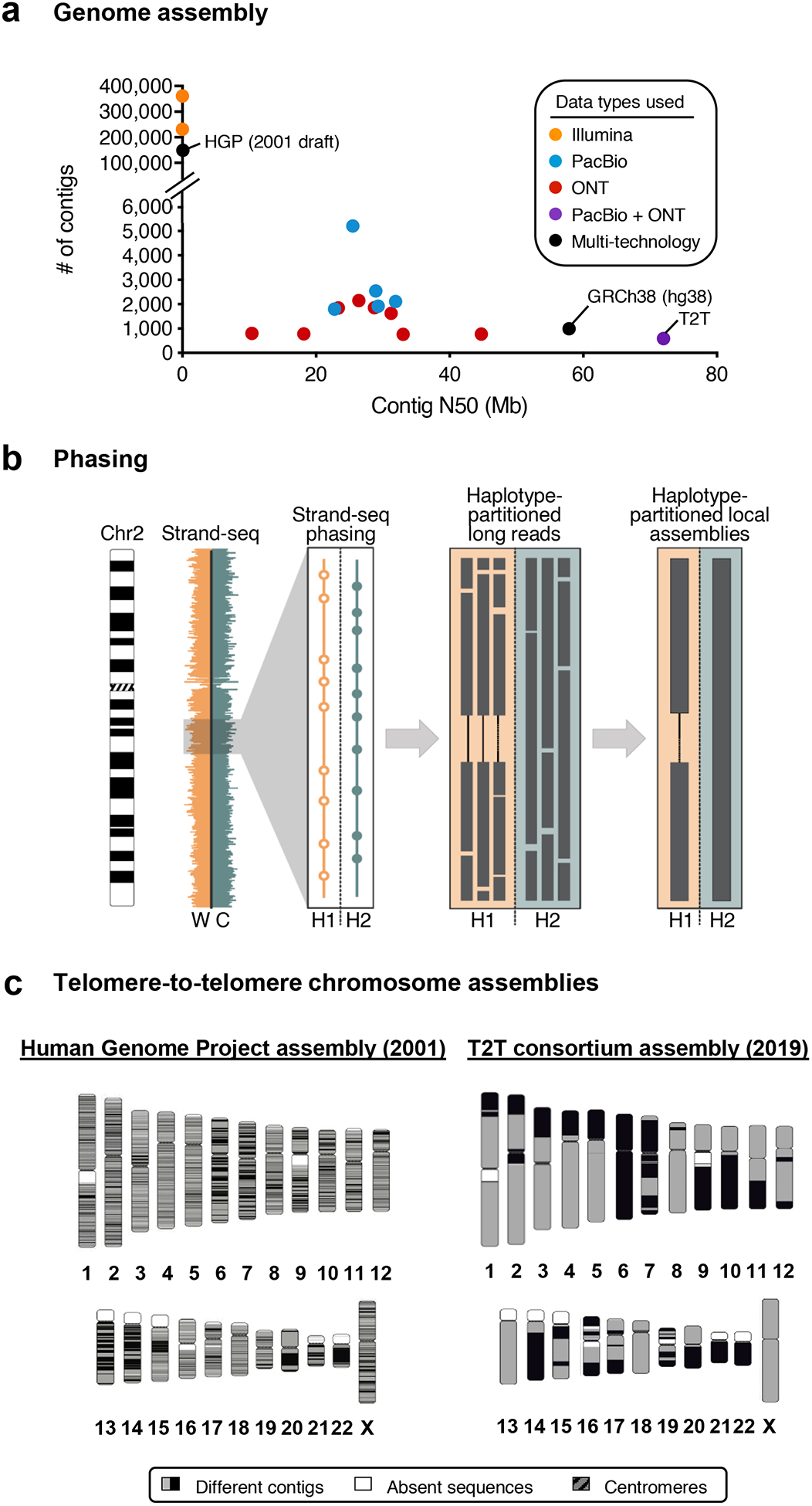
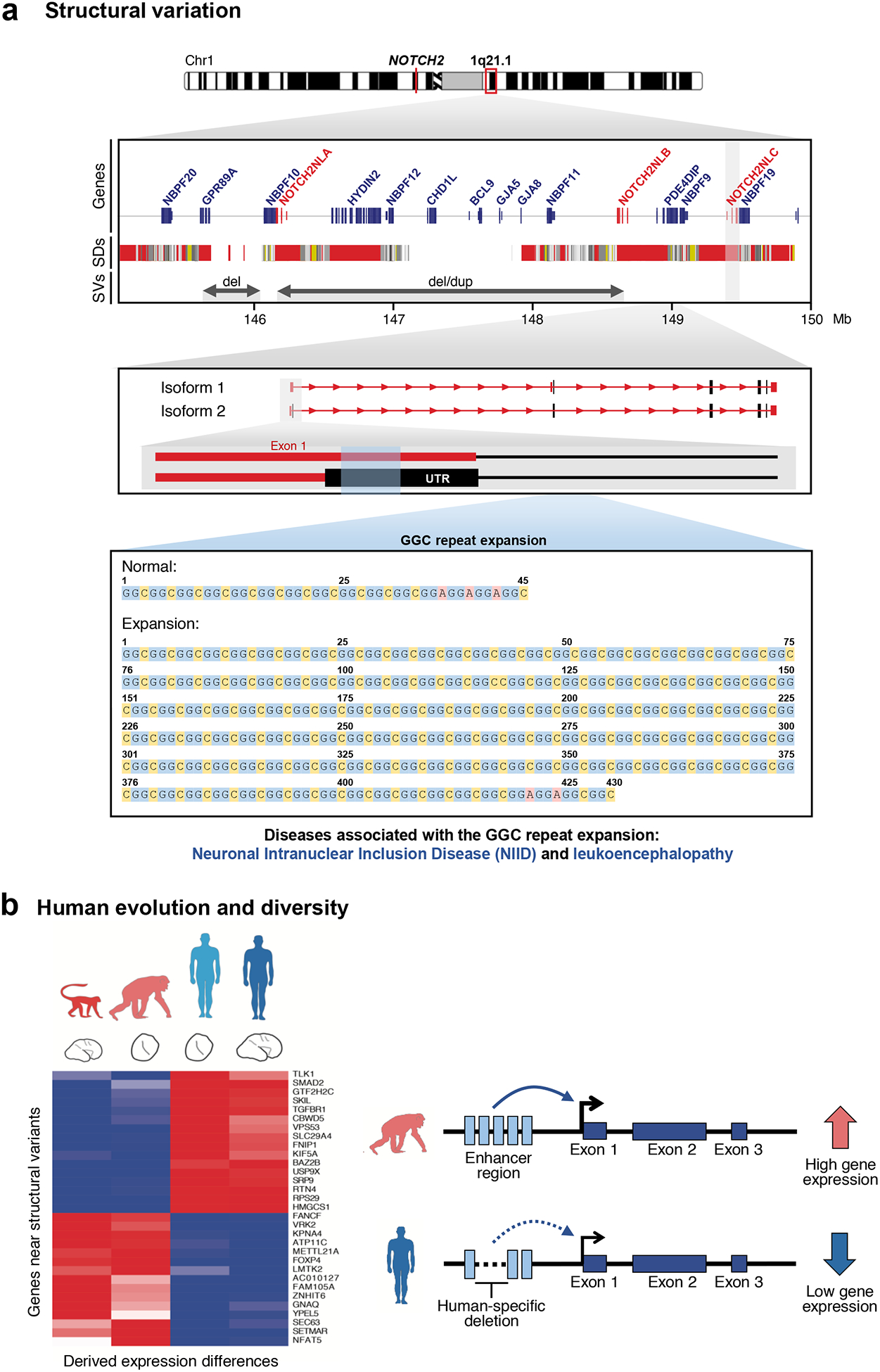
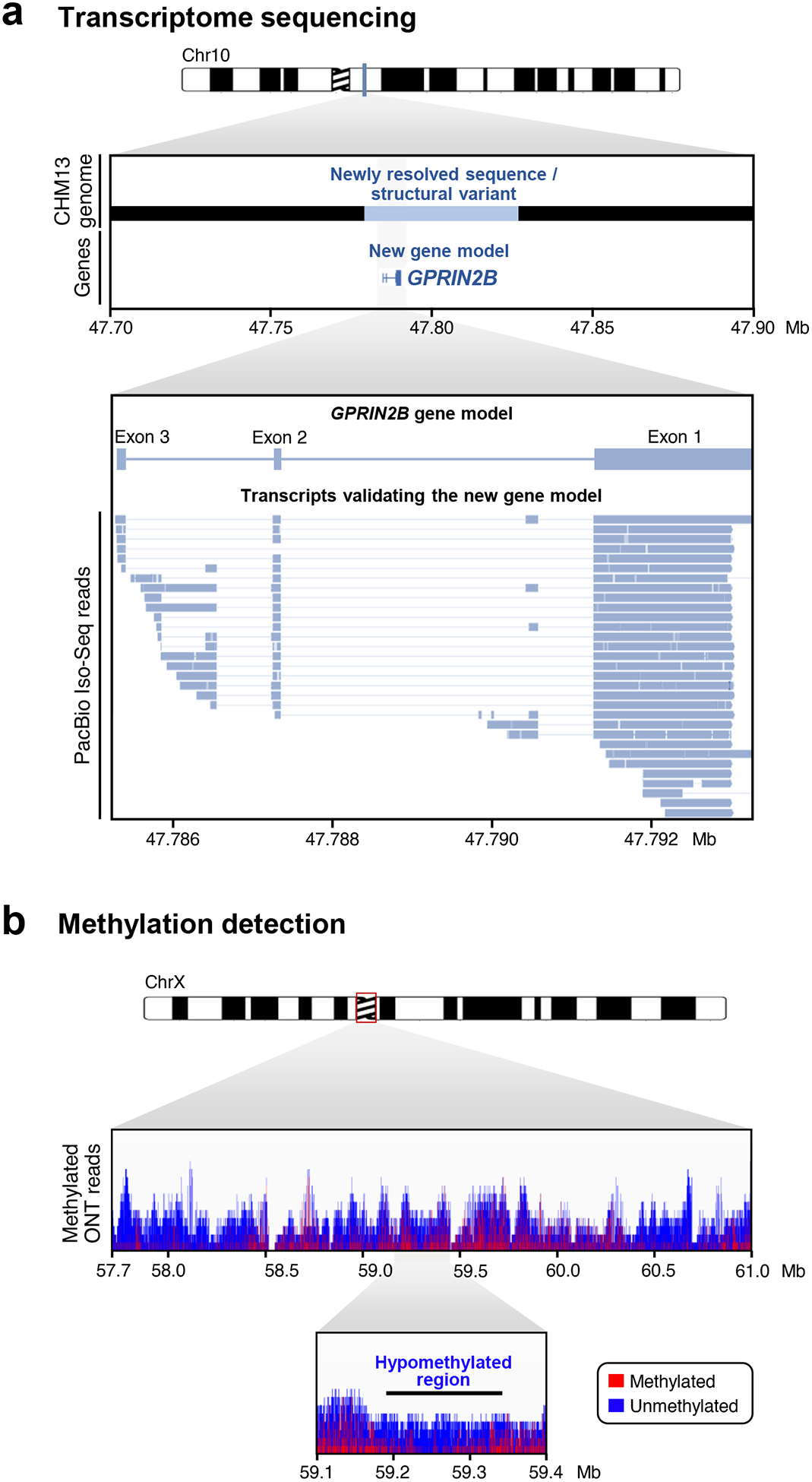
Similar articles
-
Genetic variation and the de novo assembly of human genomes.Nat Rev Genet. 2015 Nov;16(11):627-40. doi: 10.1038/nrg3933. Epub 2015 Oct 7. Nat Rev Genet. 2015. PMID: 26442640 Free PMC article. Review.
-
Leveraging the power of long reads for targeted sequencing.Genome Res. 2024 Nov 20;34(11):1701-1718. doi: 10.1101/gr.279168.124. Genome Res. 2024. PMID: 39567237 Free PMC article. Review.
-
Genomic Analysis in the Age of Human Genome Sequencing.Cell. 2019 Mar 21;177(1):70-84. doi: 10.1016/j.cell.2019.02.032. Cell. 2019. PMID: 30901550 Free PMC article. Review.
-
Semi-automated assembly of high-quality diploid human reference genomes.Nature. 2022 Nov;611(7936):519-531. doi: 10.1038/s41586-022-05325-5. Epub 2022 Oct 19. Nature. 2022. PMID: 36261518 Free PMC article.
-
ARKS: chromosome-scale scaffolding of human genome drafts with linked read kmers.BMC Bioinformatics. 2018 Jun 20;19(1):234. doi: 10.1186/s12859-018-2243-x. BMC Bioinformatics. 2018. PMID: 29925315 Free PMC article.
Cited by
-
Approaches to long-read sequencing in a clinical setting to improve diagnostic rate.Sci Rep. 2022 Oct 9;12(1):16945. doi: 10.1038/s41598-022-20113-x. Sci Rep. 2022. PMID: 36210382 Free PMC article.
-
Advances in the human skin microbiota and its roles in cutaneous diseases.Microb Cell Fact. 2022 Aug 29;21(1):176. doi: 10.1186/s12934-022-01901-6. Microb Cell Fact. 2022. PMID: 36038876 Free PMC article. Review.
-
Chromosomal translocation disrupting the SMAD4 gene resulting in the combined phenotype of Juvenile polyposis syndrome and Hereditary Hemorrhagic Telangiectasia.Mol Genet Genomic Med. 2020 Nov;8(11):e1498. doi: 10.1002/mgg3.1498. Epub 2020 Oct 15. Mol Genet Genomic Med. 2020. PMID: 33058509 Free PMC article.
-
Exome variant discrepancies due to reference-genome differences.Am J Hum Genet. 2021 Jul 1;108(7):1239-1250. doi: 10.1016/j.ajhg.2021.05.011. Epub 2021 Jun 14. Am J Hum Genet. 2021. PMID: 34129815 Free PMC article.
-
Rediscovering tandem repeat variation in schizophrenia: challenges and opportunities.Transl Psychiatry. 2023 Dec 20;13(1):402. doi: 10.1038/s41398-023-02689-8. Transl Psychiatry. 2023. PMID: 38123544 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
