

Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome
- PMID: 25024183
- PMCID: PMC4121787
- DOI: 10.1073/pnas.1411131111
Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome
Abstract
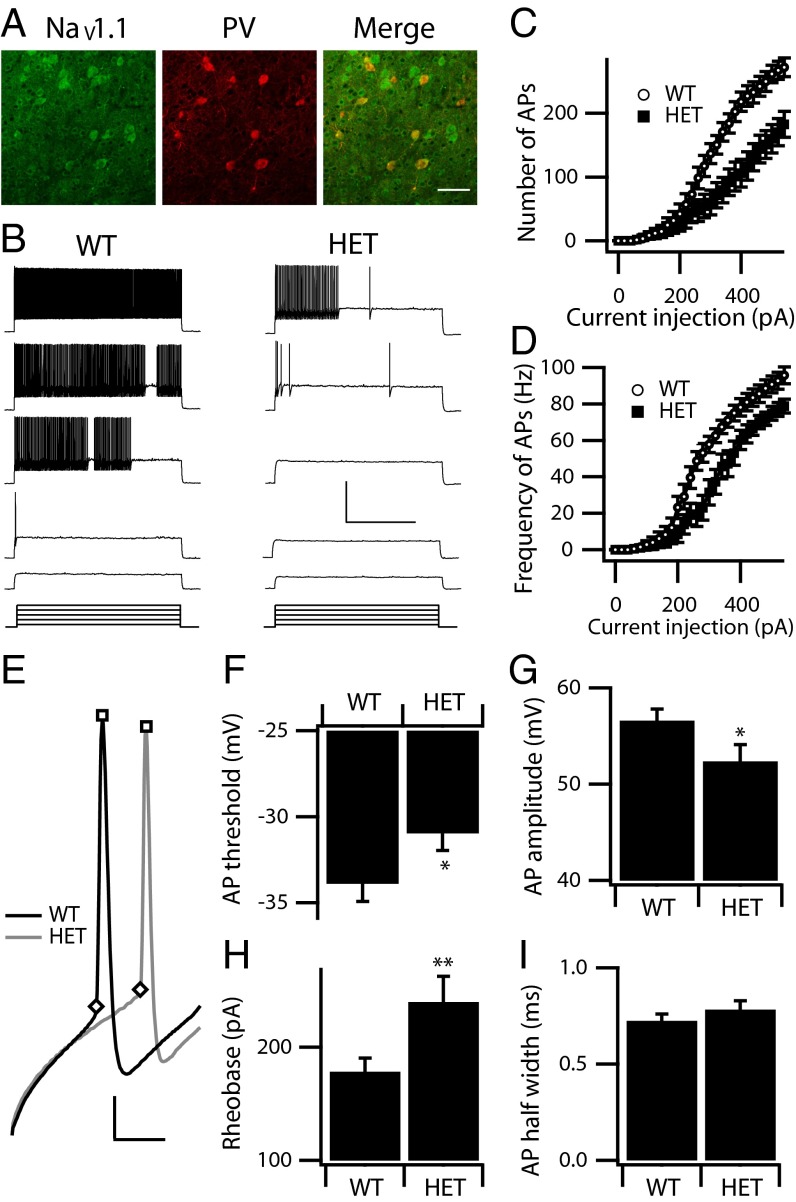
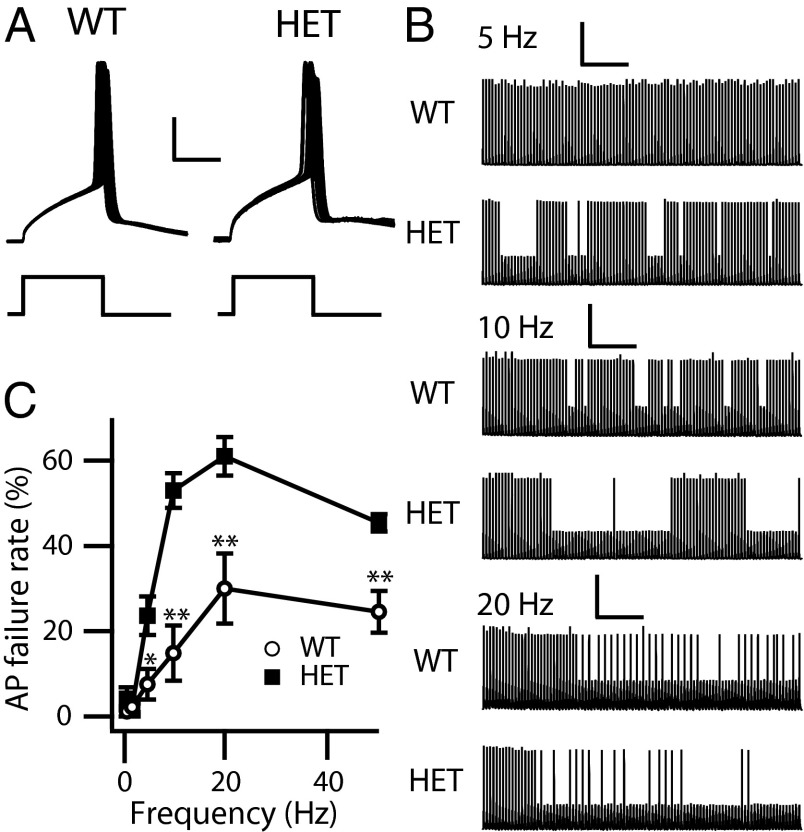
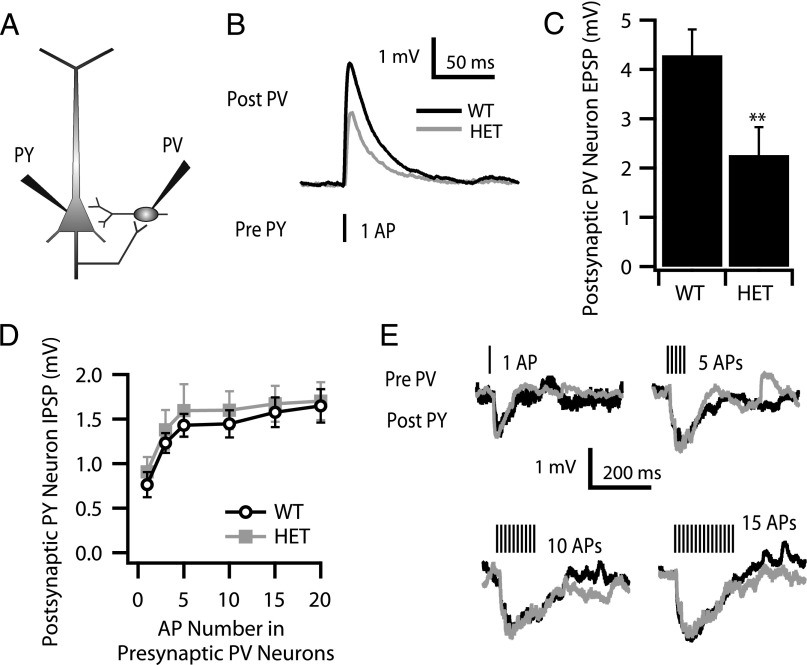
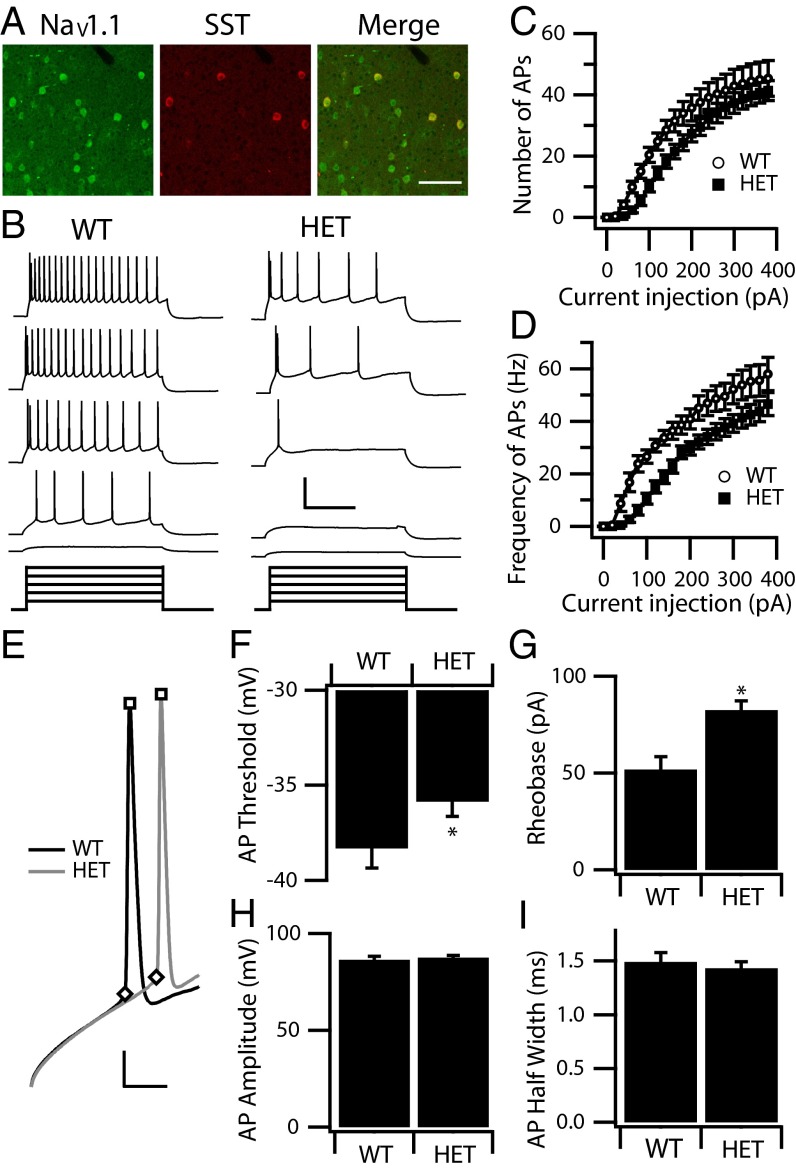
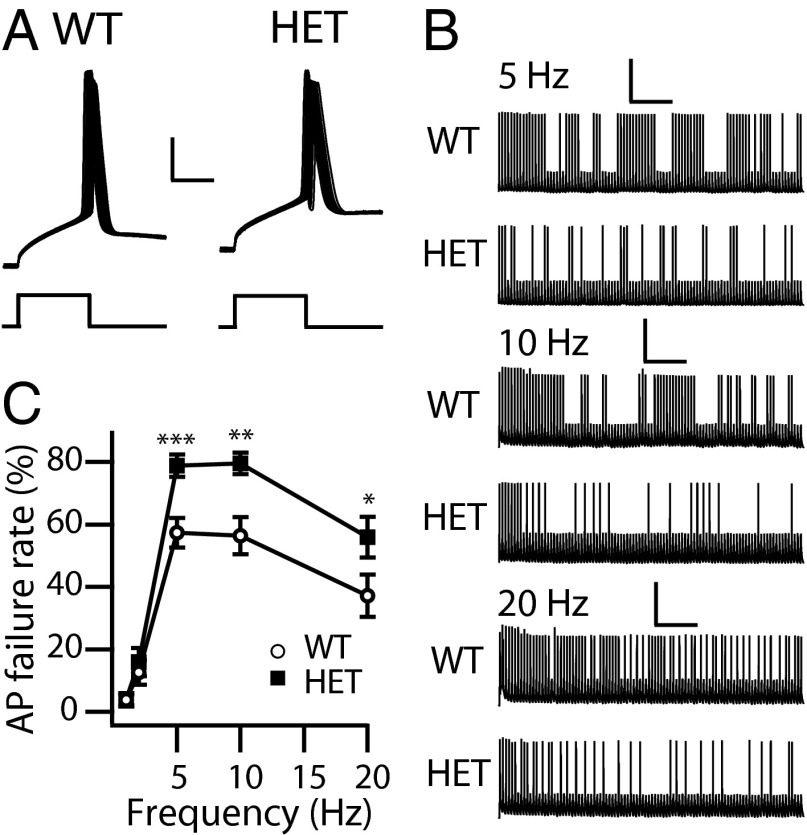
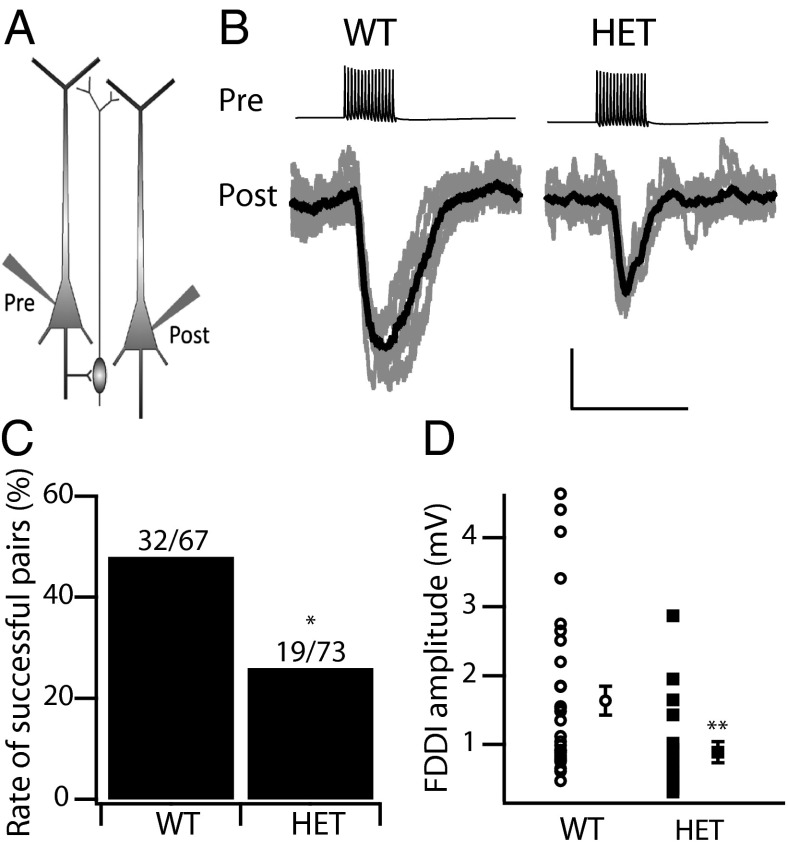
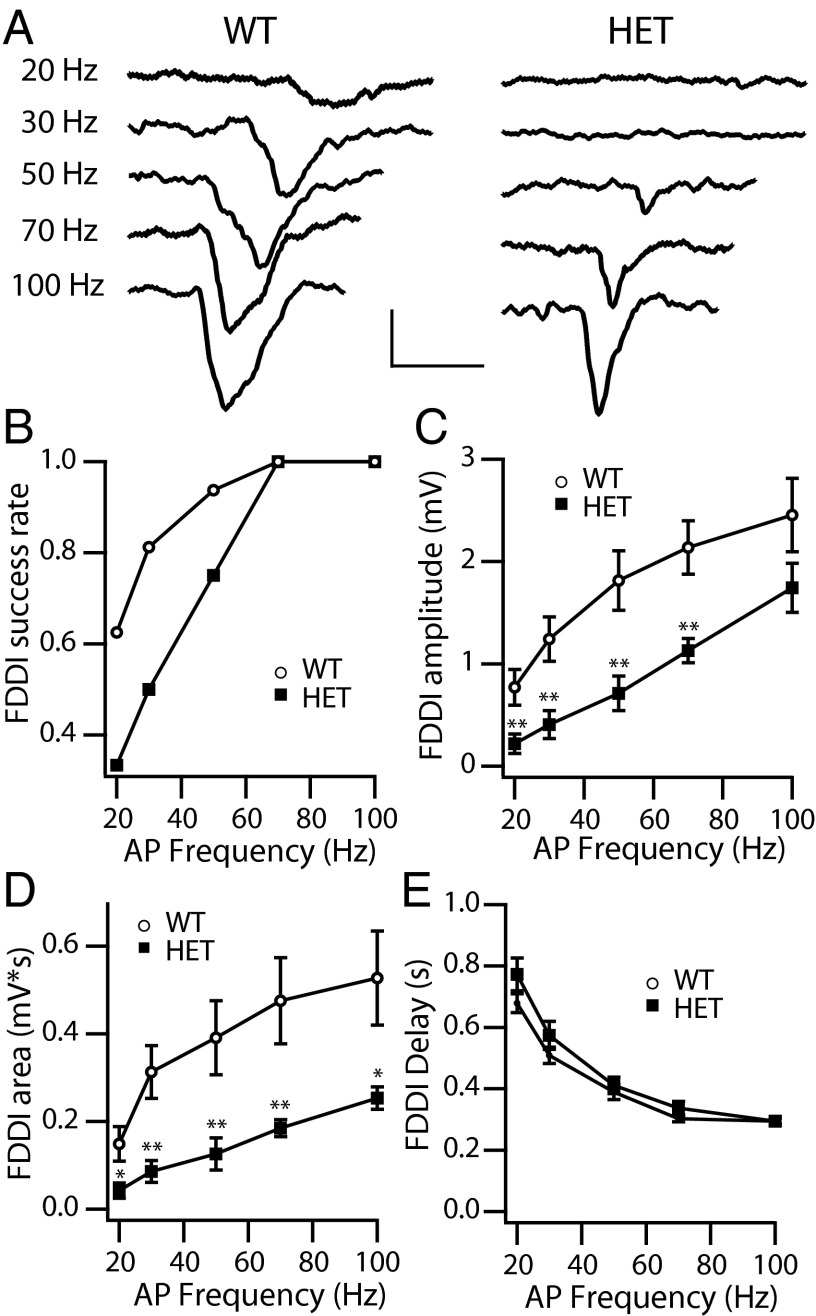
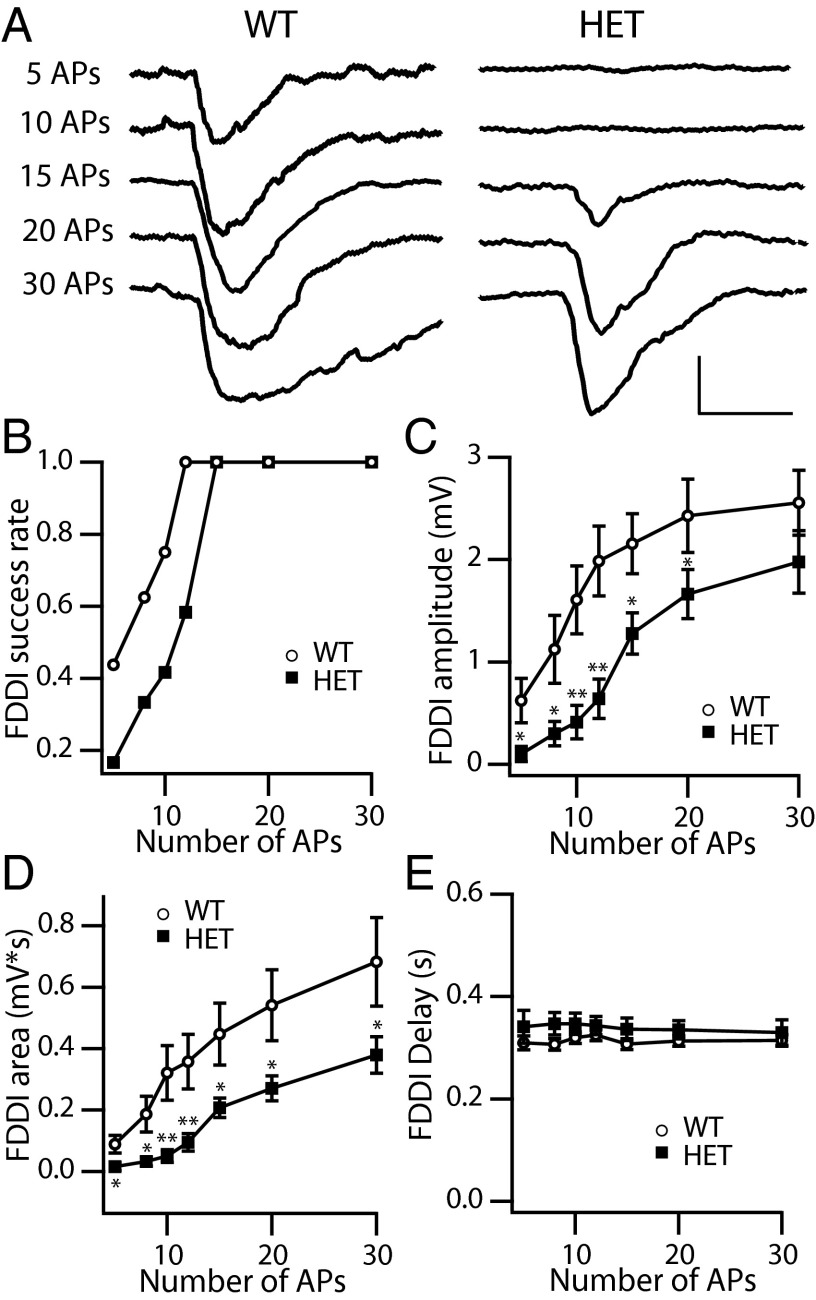
Haploinsufficiency of the voltage-gated sodium channel NaV1.1 causes Dravet syndrome, an intractable developmental epilepsy syndrome with seizure onset in the first year of life. Specific heterozygous deletion of NaV1.1 in forebrain GABAergic-inhibitory neurons is sufficient to cause all the manifestations of Dravet syndrome in mice, but the physiological roles of specific subtypes of GABAergic interneurons in the cerebral cortex in this disease are unknown. Voltage-clamp studies of dissociated interneurons from cerebral cortex did not detect a significant effect of the Dravet syndrome mutation on sodium currents in cell bodies. However, current-clamp recordings of intact interneurons in layer V of neocortical slices from mice with haploinsufficiency in the gene encoding the NaV1.1 sodium channel, Scn1a, revealed substantial reduction of excitability in fast-spiking, parvalbumin-expressing interneurons and somatostatin-expressing interneurons. The threshold and rheobase for action potential generation were increased, the frequency of action potentials within trains was decreased, and action-potential firing within trains failed more frequently. Furthermore, the deficit in excitability of somatostatin-expressing interneurons caused significant reduction in frequency-dependent disynaptic inhibition between neighboring layer V pyramidal neurons mediated by somatostatin-expressing Martinotti cells, which would lead to substantial disinhibition of the output of cortical circuits. In contrast to these deficits in interneurons, pyramidal cells showed no differences in excitability. These results reveal that the two major subtypes of interneurons in layer V of the neocortex, parvalbumin-expressing and somatostatin-expressing, both have impaired excitability, resulting in disinhibition of the cortical network. These major functional deficits are likely to contribute synergistically to the pathophysiology of Dravet syndrome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Dissecting the phenotypes of Dravet syndrome by gene deletion.Brain. 2015 Aug;138(Pt 8):2219-33. doi: 10.1093/brain/awv142. Epub 2015 May 27. Brain. 2015. PMID: 26017580 Free PMC article.
-
Antisense oligonucleotides restore excitability, GABA signalling and sodium current density in a Dravet syndrome model.Brain. 2024 Apr 4;147(4):1231-1246. doi: 10.1093/brain/awad349. Brain. 2024. PMID: 37812817 Free PMC article.
-
Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome.Neurobiol Dis. 2015 Jan;73:106-17. doi: 10.1016/j.nbd.2014.09.017. Epub 2014 Oct 2. Neurobiol Dis. 2015. PMID: 25281316 Free PMC article.
-
Sodium Channelopathies in Human and Animal Models of Epilepsy and Neurodevelopmental Disorders.In: Noebels JL, Avoli M, Rogawski MA, Vezzani A, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. 5th edition. New York: Oxford University Press; 2024. Chapter 44. In: Noebels JL, Avoli M, Rogawski MA, Vezzani A, Delgado-Escueta AV, editors. Jasper's Basic Mechanisms of the Epilepsies. 5th edition. New York: Oxford University Press; 2024. Chapter 44. PMID: 39637100 Free Books & Documents. Review.
-
Molecular and cellular basis: insights from experimental models of Dravet syndrome.Epilepsia. 2011 Apr;52 Suppl 2:70-1. doi: 10.1111/j.1528-1167.2011.03006.x. Epilepsia. 2011. PMID: 21463284 Review.
Cited by
-
Deletion of a non-canonical regulatory sequence causes loss of Scn1a expression and epileptic phenotypes in mice.Genome Med. 2021 Apr 26;13(1):69. doi: 10.1186/s13073-021-00884-0. Genome Med. 2021. PMID: 33910599 Free PMC article.
-
Neddylation stabilizes Nav1.1 to maintain interneuron excitability and prevent seizures in murine epilepsy models.J Clin Invest. 2021 Apr 15;131(8):e136956. doi: 10.1172/JCI136956. J Clin Invest. 2021. PMID: 33651714 Free PMC article.
-
Upregulation of Haploinsufficient Gene Expression in the Brain by Targeting a Long Non-coding RNA Improves Seizure Phenotype in a Model of Dravet Syndrome.EBioMedicine. 2016 Jul;9:257-277. doi: 10.1016/j.ebiom.2016.05.011. Epub 2016 May 13. EBioMedicine. 2016. PMID: 27333023 Free PMC article.
-
Use of Zebrafish Models to Boost Research in Rare Genetic Diseases.Int J Mol Sci. 2021 Dec 12;22(24):13356. doi: 10.3390/ijms222413356. Int J Mol Sci. 2021. PMID: 34948153 Free PMC article. Review.
-
GluN2A NMDA Receptor Enhancement Improves Brain Oscillations, Synchrony, and Cognitive Functions in Dravet Syndrome and Alzheimer's Disease Models.Cell Rep. 2020 Jan 14;30(2):381-396.e4. doi: 10.1016/j.celrep.2019.12.030. Cell Rep. 2020. PMID: 31940483 Free PMC article.
References
-
- Wolff M, Cassé-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): Natural history and neuropsychological findings. Epilepsia. 2006;47(Suppl 2):45–48. - PubMed
-
- Genton P, Velizarova R, Dravet C. Dravet syndrome: The long-term outcome. Epilepsia. 2011;52(Suppl 2):44–49. - PubMed
-
- Brunklaus A, Dorris L, Zuberi SM. Comorbidities and predictors of health-related quality of life in Dravet syndrome. Epilepsia. 2011;52(8):1476–1482. - PubMed
-
- Claes LR, et al. The SCN1A variant database: A novel research and diagnostic tool. Hum Mutat. 2009;30(10):E904–E920. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
